Правила GMP (Правила производства лекарственных средств)
1. Введение
1.1 Цель
Данная часть стандарта (далее – руководство) распространяется на применение Правил GMP к производству активных фармацевтических субстанций (АФС) в соответствии с системой обеспечения качества. Она также направлена на обеспечения гарантии качества и чистоты АФС в соответствии с заданными требованиями.
Термин «производство» включает в себя все виды операций с АФС: приемку исходного сырья, производство, упаковку, переупаковку, маркировку, перемаркировку, контроль качества, выпуск продукции, хранение и реализацию, а также соответствующие меры контроля. Понятия «должен», «следует», применяемые в настоящем руководстве, указывают на рекомендации, выполнение которых предполагается, за исключением случаев, когда их выполнить невозможно или когда они могут быть модифицированы в соответствии с приложениями к настоящему стандарту или они могут быть заменены альтернативными действиями, по крайней мере, с эквивалентным уровнем обеспечения качества продукции.
Руководство не затрагивает вопросов безопасности персонала, занятого в производстве, и требований по защите окружающей среды. Производитель несет ответственность за безопасность персонала и окружающей среды в соответствии с законодательством.
Настоящее руководство не устанавливает требований, предъявляемых при регистрации (подаче заявки на регистрацию) АФС, и не заменяет требований Фармакопеи, не затрагивает полномочий соответствующих органов по установлению специфических требований к АФС для выдачи разрешения на реализацию/производство или применение лекарственных средств. Следует выполнять все требования, установленные при государственной регистрации АФС.
1.2 Область применения
Настоящее руководство устанавливает требования к производству АФС, используемых в лекарственных средствах для человека и животных. К производству стерильных АФС оно применимо только до стадии стерилизации. Стандарт не распространяется на процессы стерилизации и производство стерильных АФС в асептических условиях. Эти процессы следует проводить в соответствии с требованиями настоящего стандарта (в т. ч. приведенными в приложении 1) и других нормативных документов.
В случае производства средств против эктопаразитов для применения в ветеринарии могут использоваться другие нормативные документы, обеспечивающее выполнение требований к качеству.
Данное руководство не распространяется на производство цельной крови и плазмы, производных крови и плазмы (фракционирование плазмы), но распространяется на производство АФС, получаемых с использованием крови или плазмы в качестве исходных материалов.
Настоящее руководство не распространяется на производство нерасфасованных лекарственных средств, но распространяется на активные фармацевтические субстанции, относящиеся к приложениям 2–7 настоящего стандарта.
Раздел 17 содержит требования к сторонам, которые, в числе прочего, занимаются реализацией или хранением АФС и промежуточной продукции. Эти положения рассмотрены боее подробной в правилах оптовой торговли АФС для лекарственных средств, предназначенных для человека согласно статье 47 Директивы ЕС 2001/83/ЕС.
Раздел 19 руководства содержит требования, распространяющиеся только на производство АФС, используемых для получения лекарственных средств, предназначенных для клинических исследований.
Исходный материал для производства АФС – сырье, промежуточные продукты или другие АФС, используемые в производстве АФС и включаемые в АФС в качестве существенного структурного элемента. Исходный материал для производства АФС может быть продуктом (материалом), получаемым от одного или более поставщиков, или материалом, производимым на самом предприятии. Как правило, для производства АФС используются исходные материалы с конкретными химическими свойствами и структурой.
Производитель АФС должен определить и документально оформить стадию, с которой должно начинаться производство АФС из исходного сырья. Для процессов химического синтеза эта стадия определяется как стадия ввода исходных материалов в технологический процесс производства АФС. Для других процессов (ферментации, экстракции, очистки и пр.) данную стадию определяют с учетом конкретных особенностей производства. В таблице 1 приведены стадии производства различных АФС с выделением стадий, на которых исходный материал, как правило, вводится в технологический процесс.
Начиная с этой стадии, на данные промежуточные продукты и/или стадии производства АФС действуют правила данной части стандарта. Они включают в себя требования к аттестации (испытаниям) критических стадий технологического процесса, оказывающих влияние на качество АФС. В то же время выбор стадий технологического процесса для проведения аттестации (испытаний) не обязательно означает, что эти стадии являются критическими для качества АФС.
Требования настоящей части распространяются, как правило, на стадии, выделенные в таблице 1 серым фоном. Это не означает, что в процессе производства должны выполняться все стадии, указанные в данной таблице. Строгость следования требованиям GMP должна возрастать от ранних стадий производства АФС к завершающим стадиям технологического процесса, очистки и упаковки. Физическую обработку АФС, такую как грануляция, покрытие оболочкой или физическое изменение размера частиц (например, грубый и тонкий помол), следует проводить, по крайней мере, в соответствии с требованиями настоящего стандарта.
Таблица 1 – Применение руководства к производству АФС
| Вид производства | Стадии производства АФС, на которые распространяется данное руководство (выделены серым фоном) | ||||
| Химический синтез | Производство исходного материала для АФС | Ввод исходного материала АФС в процесс | Производство промежуточного продукта | Выделение и очистка | Физическая обработка и упаковка |
| АФС, выделенные из источников животного происхождения | Получение (сбор) органа, жидкости или тканей | Измельчение, смешивание и/или первоначальная обработка | Ввод исходного материала АФС в процесс производства | Выделение и очистка | Физическая обработка и упаковка |
| АФС, выделенные из источников растительного происхождения | Сбор растений | Измельчение и первоначальная экстракция | Ввод исходного материала АФС в процесс производства | Выделение и очистка | Физическая обработка и упаковка |
| Растительные экстракты, используемые в качестве АФС | Сбор растений | Измельчение и первоначальная экстракция | — | Дальнейшая экстракция | Физическая обработка и упаковка |
| АФС, состоящие из размельченных или растертых в порошок растений | Сбор растений
и/или культивирование и сбор |
Измельчение (растирание) | — | — | Физическая обработка и упаковка |
| Биотехнология: ферментация/культура клеток | Создание главного и рабочего банков клеток | Поддерживание рабочего банка клеток | Культура клеток и/или ферментация | Выделение и очистка | Физическая обработка и упаковка |
| «Классическая» ферментация для производства АФС | Создание банка клеток | Поддерживание банка клеток | Ввод клеток в процесс ферментации | Выделение и очистка | Физическая обработка и упаковка |
| УСИЛЕНИЕ ТРЕБОВАНИЙ GMP → | |||||
Настоящий стандарт не распространяется на технологические стадии, предшествующие вводу исходных материалов для производства АФС в технологический процесс.
Термин «активная фармацевтическая субстанция» может использоваться наряду с термином
«активная субстанция». Термины и определения, приведенные в разделе 20 Части II относятся только к этой части. Ряд терминов определен в части I и должен использоваться в том же смысле, что и в Части I.
2. Обеспечение качества
2.1 Общие положения
-
- Ответственность за качество выпускаемого продукта несет весь персонал предприятияпроизводителя.
- На каждом предприятии следует разработать, документально оформить и внедрить систему обеспечения качества, которая предусматривает активное участие в ней руководителей предприятия и всего персонала, занятого в производстве.
- Система обеспечения качества охватывает организационную структуру, инструкции, методики, процессы и ресурсы, а также действия, необходимые для обеспечения гарантии соответствия АФС всем требованиям к качеству и чистоте. Деятельность, связанная с обеспечением качества,должна быть четко определена и документально оформлена.
-
- Подразделения (отделы, службы), ответственные за обеспечение качества и контроль качества, должны быть независимыми от производства. Эти функции могут выполнять отдельные подразделения по обеспечению и контролю качества либо быть возложены на одно лицо (группу лиц) в зависимости от объемов и структуры предприятия.
- Следует определить круг лиц, ответственных за выпуск промежуточных продуктов и АФС.
- Все действия по контролю качества следует оформлять документально непосредственно при их выполнении.
- Любые отклонения от принятых инструкций должны быть обоснованы и оформлены документально. Критические отклонения следует расследовать с необходимым документальным оформлением.
- Не допускается выпускать или использовать материалы до получения положительного заключения отдела(ов) качества, если не установлен специальный порядок, допускающий соответствующие отклонения (например, выпуск материалов, находящихся в карантине, по п. 10.20 настоящего руководства, или использование сырья или промежуточных продуктов, оценка качества которых еще не завершена).
- Следует разработать порядок своевременного извещения руководства предприятия о результатах инспекций, проводимых органами контроля и надзора, случаях серьезного нарушения требований настоящего стандарта, обнаружения несоответствия продукта и принятых мерах (претензиях к качеству продукции, отзывах продукции, заключениях инспекции и т.д.).
- Для надежного обеспечения качества следует разработать и применять систему качества, включающую настоящий стандарт (GMP), контроль качества и анализ рисков.
2.2 Анализ рисков для качества
-
- Анализ рисков является систематизированным процессом оценки, контроля, сопоставления и повторного рассмотрения рисков для качества АФС. Он может выполняться к в перспективном, так и ретроспективном плане.
- Система анализа рисков должна обеспечивать:
- оценку рисков для качества, которая основана на научных знаниях и опыте работы с процессами и, в конечном итоге, на защиту пациента путем взаимодействия с потребителем АФС;
- уровни усилий, формализации и документального оформления анализа рисков зависят от степени риска.
Примеры применения методов анализа рисков могут быть найдены, помимо прочего в Части III настоящего стандарта.
2.3 Функции и ответственность подразделений по обеспечению и контролю качества
-
- Деятельность подразделений по обеспечению и контролю качества распространяется на все аспекты качества.
- Подразделения по обеспечению и контролю качества должны рассматривать и согласовывать (утверждать) все документы, имеющие отношение к качеству продукции.
- Основные функции и ответственность этих независимых подразделений качества не могут быть переданы другим подразделениям или лицам. Функции и ответственность этой службы должны быть оформлены документально и включать в себя, по крайней мере, следующее:
- Выпуск или отзыв любых АФС. Выпуск или отзыв промежуточных продуктов, используемых вне зоны контроля предприятия-производителя.
- Разработку и ввод в действие системы выпуска или отзыва сырья, промежуточных продуктов, упаковочных и печатных материалов (материалов для маркировки).
- Проверки протоколов на серии готовой продукции и протоколов лабораторных анализов на критических стадиях технологического процесса до выпуска АФС в реализацию.
- Контроль за расследованием причин критических отклонений параметров от установленных значений с принятием необходимых мер.
- Согласование всех спецификаций и технологических инструкций по производству продукции.
- Согласование всех инструкций и методик по качеству промежуточных продуктов или АФС.
- Контроль проведения внутренних проверок (самоинспекций).
- Согласование производителей промежуточных продуктов и АФС, работающих по контракту.
- Согласование изменений, которые могут оказать влияние на промежуточные продукты и АФС.
- Рассмотрение и согласование протоколов и отчетов о проведении аттестации (испытаний).
- Контроль расследования претензий, связанных с качеством продукции, и принятия необходимых мер.
- Контроль своевременного выполнения технического обслуживания и калибровки (поверки) критического оборудования в соответствии с установленным порядком.
- Контроль проведения испытаний и оформления их результатов.
- Контроль (при необходимости) данных о стабильности АФС и/или промежуточных продуктов для обоснования даты их повторного контроля, срока годности и условий их хранения.
- Составление отчетов о качестве продукции (подраздел 2.5 настоящего руководства).
2.4 Функции и ответственность производственных подразделений
Функции и ответственность производственных подразделений должны быть оформлены документально и включать в себя, по крайней мере, следующее:
- Разработку, пересмотр и согласование (утверждение) инструкций по производству промежуточных продуктов и/или АФС, а также обеспечение инструкциями всех причастных подразделений и исполнителей в соответствии с утвержденной инструкцией.
- Производство АФС и, при необходимости, промежуточных продуктов в соответствии с утвержденными инструкциями.
- Рассмотрение всех протоколов производства серии продукции и подтверждение их полноты и правильности оформления (подписания) в установленном порядке.
- Контроль проведения анализа всех отклонений в процессе производства и расследований критических отклонений с соответствующим документальным оформлением.
- Контроль чистоты производственных помещений и, при необходимости, их дезинфекции.
- Контроль выполнения необходимых калибровок (поверок) и хранения протоколов калибровки.
- Контроль содержания помещений и оборудования в надлежащем состоянии и хранения соответствующих протоколов.
- Контроль ведения и согласования (утверждения) протоколов и отчетов аттестации (испытаний) оборудования и процессов.
- Оценка предлагаемых изменений в продукции, технологическом процессе или оборудовании.
- Контроль проведения аттестации новых и, при необходимости, модернизированных помещений и оборудования.
2.5 Внутренние аудиты (самоинспекции)
-
- Для подтверждения соответствия производства АФС требованиям настоящего стандарта следует регулярно проводить внутренние аудиты согласно утвержденному графику.
- Результаты аудита и последующие корректирующие действия следует оформлять документально и представлять руководству предприятия. Действия по результатам аудита следует выполнять своевременно и надлежащим образом.
2.6 Анализ качества продукции
-
- Для подтверждения постоянного соответствия технологического процесса установленным требованиям следует регулярно проводить анализ качества АФС. Такой анализ следует проводить, как правило, ежегодно с последующим документальным оформлением. Отчет о проведении анализа качества продукции должен включать в себя, по крайней мере, следующее:
- рассмотрение результатов внутрипроизводственного контроля и испытаний АФС по критическим параметрам;
- анализ данных о всех сериях продукции, качество которых не соответствовало установленным требованиям;
- анализ всех существенных отклонений в технологическом процессе и результаты расследования их причин;
- рассмотрение всех изменений, внесенных в технологический процесс или аналитические методы;
- рассмотрение результатов контроля стабильности;
- анализ всех претензий, возвратов и отзывов, связанных с качеством продукции;
- анализ эффективности всех корректирующих действий.
-
- Следует оценивать результаты анализа и принять решение о необходимости принятия соответствующих мер или повторной аттестации (испытаний). Обоснование необходимости этих мер должно быть оформлено документально. Указанные меры должны быть выполнены своевременно и надлежащим образом.
3. Персонал
3.1 Квалификация персонала
-
- Производство должно быть укомплектовано необходимым числом сотрудников, имеющих соответствующее образование, прошедших обучение и/или имеющих достаточный опыт для работы в производстве АФС и промежуточных продуктов.
- Ответственность и обязанности всех сотрудников, работающих в производстве АФС и промежуточных продуктов, должны быть документально оформлены.
- Следует проводить регулярное обучение персонала с привлечением специалистов необходимой квалификации. Это обучение должно охватывать, как минимум, вопросы, связанные с выполнением конкретных операций и требований настоящего стандарта в части обязанностей данного сотрудника. Следует вести и сохранять протоколы обучения сотрудников, а также проводить периодическую оценку их квалификации.
3.2 Гигиена персонала
-
- Персонал должен соблюдать правила гигиены.
- Персонал должен носить чистую одежду, соответствующую его производственной деятельности. Следует определить порядок замены этой одежды. Для защиты АФС и промежуточных материалов от загрязнения, при необходимости, следует носить дополнительные элементы одежды, покрывающие голову, лицо и руки.
- Следует избегать прямого контакта персонала с промежуточными продуктами и АФС.
- Курение, прием пищи и напитков, жевание резинки и хранение продуктов питания допускаются только в специально выделенных местах, отделенных от производственных помещений.
- Сотрудники с инфекционными заболеваниями или имеющие открытые повреждения на незакрытых участках тела не допускаются к работе, если это может отразиться на качестве АФС. Любой сотрудник с заболеванием или наличием открытой раны, выявленными при медицинском осмотре или наблюдении, должен быть отстранен от производственных операций, при выполнении которых состояние его здоровья может неблагоприятно повлиять на качество АФС, до выздоровления или получения медицинского заключения о том, что нахождение этого сотрудника на рабочем месте не представляет риска для безопасности или качества АФС.
3.3 Консультанты
-
- Консультанты по вопросам производства и контроля качества промежуточных материалов или АФС должны иметь соответствующее образование, подготовку и опыт работы или сочетание вышеперечисленных требований.
- Следует организовать ведение и хранение протоколов о проведении консультаций с указанием имен, адресов, специальностей и видов услуг, предоставляемых этими консультантами.
4. Здания, помещения и инженерные системы
4.1 Проектирование и строительство
-
- При размещении, проектировании и строительстве зданий и помещений, предназначенных для производства промежуточных продуктов и АФС, следует предусматривать удобство их эксплуатации и обслуживания в соответствии с видом и стадией производства. При проектировании помещений следует сводить к минимуму возможность загрязнения. Если для промежуточных продуктов и АФС установлены требования к микробиологической чистоте, то при проектировании помещений следует предусматривать защиту от микробных загрязнений.
- Здания и помещения должны иметь достаточные размеры для размещения оборудования и материалов с целью предотвращения их перепутывания и загрязнения.
- Оборудование, обеспечивающее надлежащую защиту материалов (например, закрытая или изолированная система), может выходить за пределы производственного помещения.
- При организации потоков материалов и персонала в зданиях или помещениях должно быть предусмотрено предотвращение перепутывания и загрязнения материалов и продукции.
- Следует предусматривать отдельные зоны или другие адекватные решения для следующих операций:
-
- получения, идентификации, отбора проб и нахождения в карантине исходных материалов до выдачи разрешения на выпуск или отклонение;
- нахождения в карантине до выпуска или отклонения промежуточных продуктов или АФС;
- отбора проб промежуточных продуктов или АФС;
- хранения отклоненных материалов до принятия решения об их дальнейшем использовании (например, при возврате, переработке или уничтожении);
-
- хранения выпущенных материалов;
- выполнения технологических операций;
- выполнения операций по упаковке и маркировке;
- проведения лабораторных анализов.
-
- Следует предусматривать необходимые помещения для подготовки персонала (мытье рук и пр.) и туалеты, обеспечить их горячей и холодной водой, мылом или другими моющими средствами, фенами для рук или одноразовыми полотенцами. Помещения для подготовки персонала и туалеты должны быть отделены от производственных зон (не иметь в них прямого выхода), но должны быть легкодоступными для персонала. При необходимости следует предусмотреть помещения для принятия душа и/или переодевания.
- Как правило, лаборатории и помещения, в которых проводят анализы, должны быть расположены отдельно от производственных зон. Если проводимые в зонах технологические операции не влияют на результаты анализа, а работа лабораторий не оказывает отрицательного влияния на производство промежуточных продуктов и АФС, лаборатории (в частности, лаборатории внутрипроизводственного контроля) могут быть расположены в производственных зонах.
4.2 Инженерные системы
-
- Следует проводить аттестацию инженерного оборудования и технологических сред (например, пара, газов, сжатого воздуха, систем отопления, вентиляции, кондиционирования и пр.), которые могут оказать влияние на качество продукции, и обеспечить их контроль и техническое обслуживание. При несоответствии параметров установленным значениям следует принимать необходимые меры. На предприятии должен храниться соответствующий комплект документации.
- При необходимости следует предусматривать системы вентиляции, очистки и вытяжки воздуха. При проектировании и мониторинге этих систем следует свести к минимуму риск прямого и перекрестного загрязнения и предусмотреть соответствующий контроль параметров на каждой технологической стадии (например, давления воздуха, наличия микроорганизмов (при необходимости), запыленности, влажности и температуры). Особое внимание следует уделить зонам, в которых АФС подвергаются воздействию окружающей среды.
- В производственных помещениях с рециркуляцией воздуха следует предусмотреть меры по предотвращению риска прямого и перекрестного загрязнения.
- Следует предусмотреть маркировку или иной метод идентификации стационарных систем трубопроводов (например, путем маркировки отдельных трубопроводов, обеспечением документации, применением систем с компьютерным управлением и контролем и пр.). Расположение системы трубопроводов не должно представлять опасность загрязнения промежуточных продуктов и АФС.
- Канализационные сливы должны иметь соответствующие размеры и, при необходимости, иметь разрыв струи либо другие средства предотвращения обратного потока.
4.3 Подготовка воды
-
- Вода, используемая при производстве АФС, должна соответствовать своему назначению, что должно быть подтверждено документально.
- За исключением отдельно оговоренных случаев, качество воды, используемой в производстве, должно, как минимум, соответствовать требованиям действующих стандартов, предъявляемым к питьевой воде.
- Если для обеспечения качества АФС характеристик питьевой воды недостаточно и необходимы более жесткие требования к химическим и/или микробиологическим характеристикам воды, должны быть разработаны требования к воде по физико-химическим свойствам, общему числу микроорганизмов, числу нежелательных микроорганизмов и/или содержанию эндотоксинов в воде.
- Следует проводить аттестацию процесса подготовки воды определенного качества, используемой в технологическом процессе, и контролировать ее параметры с установлением необходимых пределов действия.
- Если нестерильные АФС предназначены для дальнейшего производства стерильных лекарственных средств (или их производитель заявляет о пригодности АФС для этой цели), то вода, применяемая на конечных стадиях выделения и очистки, должна быть проверена на общее число микроорганизмов, число нежелательных микроорганизмов и уровень эндотоксинов.
4.4 Разделение зон
-
- Для производства продукции с высокой сенсибилизирующей активностью (пенициллины или цефалоспорины и пр.) следует использовать специально выделенные производственные зоны, в состав которых могут входить помещения, вентиляционное и/или технологическое оборудование.
- Специальные производственные площади должны быть выделены для производства про-
дукции с инфицирующими свойствами, высокой фармакологической активностью или токсичностью (например, некоторых стероидов, цитотоксических противоопухолевых средств), за исключением случаев, когда методы инактивации и/или очистки оборудования прошли аттестацию (испытания) и выполняются в установленном порядке.
-
- Следует предусмотреть меры по предотвращению перекрестного загрязнения при перемещении персонала, материалов и т.д. из одной выделенной зоны в другую.
- Не допускается проводить в зданиях и/или на оборудовании, предназначенном для производства АФС, любые производственные операции (в т. ч. взвешивание, размол или упаковку) с высокотоксичными нефармацевтическими материалами, например, гербицидами и пестицидами. Обращение с высокотоксичными нефармацевтическими материалами и их хранение должны быть отделены от АФС.
4.5 Освещение
4.50 Для облегчения правильной эксплуатации, обслуживания и очистки помещений следует предусмотреть необходимый уровень их освещения.
4.6 Стоки и отходы
4.60 Стоки и отходы (например, твердые, жидкие или газообразные побочные продукты производства) должны своевременно и безопасно удаляться из зданий и окружающей их зоны с соблюдением санитарных норм. Контейнеры и/или трубопроводы для отходов должны иметь четкую маркировку.
4.7 Уборка, дезинфекция и техническое обслуживание
-
- Здания и помещения, в которых производятся промежуточные продукты и АФС, должны соответствующим образом обслуживаться, ремонтироваться и содержаться в чистоте.
- Для проведения уборки (обработки, дезинфекции) должны быть разработаны специальные инструкции с указанием используемого оборудования, материалов и графиков выполнения работ.
- При необходимости, для предотвращения загрязнения оборудования, сырья, упаковочных и печатных материалов, промежуточных продуктов и АФС следует разработать инструкции по борьбе с грызунами, использованию инсектицидов, фунгицидов, фумигирующих средств, а также чистящих и дезинфицирующих средств.
5. Технологическое оборудование
5.1 Требования к конструкции и монтажу
-
- Оборудование, применяемое в производстве промежуточных продуктов и АФС, должно иметь соответствующую конструкцию и размеры и располагаться в месте, удобном для его использования, очистки, дезинфекции и технического обслуживания.
- Поверхности оборудования, контактирующие с сырьем, промежуточными продуктами или АФС, не должны влиять на качество промежуточных продуктов и АФС и изменять их характеристики за пределы допустимых значений.
- Технологическое оборудование должно использоваться только по назначению.
- Основное оборудование и стационарные технологические линии (реакторы, контейнеры для хранения, технологические линии и пр.), используемые в производстве промежуточных продуктов и АФС, должны иметь соответствующую маркировку.
- Не допускается контакт всех веществ, применяемых в технологическом оборудовании (смазок, нагревающих и охлаждающих жидкостей) с промежуточными продуктами и АФС, который может привести к изменению качества промежуточных продуктов и АФС за пределы установленных требований. Необходимо расследовать любые отклонения от этих требований, чтобы не допускать отрицательного влияния вспомогательных веществ на перерабатываемые материалы. По возможности следует использовать смазки и масла, предназначенные для пищевой промышленности.
- Оборудование, по возможности, должно быть закрытым или герметичным. При использовании открытого оборудования необходимо принимать меры по снижению риска загрязнения до минимума.
- Следует хранить действующий комплект документации на оборудование и установки (например, контрольно-измерительные приборы, вспомогательные системы и пр.).
5.2 Техническое обслуживание и очистка оборудования
-
- Техническое обслуживание оборудования следует проводить в соответствии с утвержденными графиками и инструкциями, в которых должны быть указаны ответственные лица.
-
- Следует разработать письменные инструкции по очистке оборудования и его допуску к последующему использованию в производстве промежуточной продукции и АФС. Эти инструкции должны быть достаточно подробными, чтобы операторы могли выполнить очистку каждого единицы оборудования эффективно и в неизменном порядке. Инструкции должны содержать следующие данные:
-
- определение ответственности за очистку оборудования;
- график проведения очистки и, при необходимости, дезинфекции;
- подробное описание методов и материалов, в т. ч. приготовления моющих средств;
- инструкции по разборке и сборке каждого узла оборудования (при необходимости);
- порядок удаления и уничтожения данных о предыдущей серии продукции;
- инструкции по предохранению чистого оборудования от загрязнения до начала его использования;
- порядок проверки чистоты оборудования непосредственно перед его использованием (при необходимости);
- максимально допустимое время после окончания производства до начала очистки оборудования (при необходимости).
-
- Для предотвращения прямого и перекрестного загрязнений или переноса материалов, которые могут повлиять на соответствие качества промежуточных продуктов и АФС установленным требованиям, следует предусматривать надлежащее хранение, очистку оборудования и принадлежностей (при необходимости, дезинфекцию или стерилизацию).
- Для оборудования, предназначенного для непрерывного производства или производства циклами серий одних и тех же промежуточных продуктов или АФС, следует предусмотреть его очистку через определенные интервалы времени для предотвращения накопления и переноса загрязнений (например, продуктов распада или недопустимой концентрации микроорганизмов).
- Для неспециализированного оборудования между производством различных материалов следует предусмотреть его очистку с целью предупреждения перекрестного загрязнения.
- Должны быть установлены допустимые предельные значения остатков загрязнений, а также методы и материалы, необходимые для очистки.
- Проведение очистки оборудования должно быть документально оформлено. Оборудование должно иметь соответствующую маркировку.
5.3 Калибровка (поверка)
-
- Калибровку (поверку) контрольно-измерительного и аналитического оборудования (в т. ч. весов), имеющего критическое значение для обеспечения качества промежуточных продуктов или АФС, следует проводить в плановом порядке.
- Калибровку (поверку) оборудования следует проводить с использованием стандартных образцов (эталонов), прослеживаемых до соответствующих эталонных образцов (если они существуют).
- Протоколы проведения калибровок (поверок) должны сохраняться.
- Данные о состоянии калибровки (поверки) критического оборудования должны находиться в доступном месте.
- Не допускается использовать приборы, не соответствующие требованиям калибровки (поверки).
- При обнаружении отклонений в работе контрольно-измерительного оборудования от установленных требований следует выяснить, могут ли они оказать влияние на качество промежуточных продуктов или АФС, произведенных с использованием этого оборудования после последнего проведения калибровки (поверки).
5.4 Системы с компьютерным управлением и контролем
-
- Системы с компьютерным управлением и контролем, относящиеся к выполнению требований GMP, должны быть аттестованы (испытаны) с учетом разнообразия, сложности и критичности использования компьютеров.
- Пригодность систем с компьютерным управлением и контролем и программного обеспечения для выполнения поставленной задачи должна быть показана при их аттестации (испытаниях) в установленном и оснащенном состояниях.
- Если программное обеспечение аттестовано до его получения, то не требуется проводить его проверку в аналогичном объеме. Если система с компьютерным управлением и контролем не была аттестована (испытана) при монтаже, то при наличии необходимой документации может быть проведена ее ретроспективная аттестация (испытания).
- Следует предусмотреть защиту систем с компьютерным управлением и контролем от несанкционированного доступа или изменения данных, а также защиту от потери данных (например, при выключении компьютера).
Должна регистрироваться информация о любых изменениях данных, последнем вводе данных,
- том, кем и когда они были сделаны.
-
- Работа и обслуживание систем с компьютерным управлением и контролем должны проводиться в соответствии с инструкциями.
- При вводе существенной информации вручную следует предусмотреть дополнительную проверку правильности ввода данных. Такая проверка может выполняться другим оператором либо самой системой.
- Сбои в работе систем с компьютерным управлением и контролем, которые могут оказать влияние на качество промежуточных продуктов или АФС, на надежность записей или результатов испытаний, следует регистрировать и расследовать.
- Внесение изменений в систему с компьютерным управлением и контролем должно проводиться в соответствии с методикой внесения изменений, оформляться документально и утверждаться ответственным лицом с проверкой внесения этих изменений. Следует хранить протоколы всех изменений и усовершенствований, внесенных в компьютер, программное обеспечение или любой другой критический элемент системы с компьютерным управлением и контролем.
- Если сбои или отказы в работе системы могут привести к необратимой потере данных, следует предусмотреть резервную систему. Для всех систем с компьютерным управлением и контролем должны быть предусмотрены меры, гарантирующие защиту данных.
- Допускается запись информации с помощью других средств.
6. Документация и протоколы
6.1 Система документации и спецификации
-
- Разработку, пересмотр, утверждение и распространение всех документов, относящихся к производству промежуточных продуктов или АФС, следует выполнять по специальным инструкциям (методикам). Эти документы могут быть оформлены как в письменном, так и в электронном виде.
- Выпуск, пересмотр, замена и отмена всех документов должны контролироваться с сохранением сведений об их предыдущих версиях.
- Следует организовать систему хранения всех необходимых документов (отчетов о развитии производства, объемах производства, замене оборудования и аттестации технологических процессов, протоколов обучения персонала, выпуска продукции, контроля качества и реализации продукции и пр.) и установить сроки хранения этих документов.
- Вся документация о производстве, контроле качества и реализации продукции должна храниться не менее одного года после окончания срока годности данной серии. Для АФС с установленной датой повторного контроля срок хранения документов — не менее трех лет со дня полной реализации серии.
- Записи в протоколы следует вносить в предназначенные для этого места сразу же после выполнения действий с указанием ответственного лица, сделавшего эти записи, так, чтобы их нельзя было удалить. Исправления в записях должны быть внесены таким образом, чтобы можно было прочесть первоначальную запись. Исправления должны содержать дату внесения изменений и подпись ответственного лица.
- Оригиналы или копии протоколов, регистрирующие какие-либо действия, следует хранить в соответствующих подразделениях в течение всего срока хранения документации. Допускается хранить протоколы в других местах, если используется электронный или любой другой способ переноса информации.
- Спецификации, инструкции, методики и протоколы могут храниться в виде оригиналов или заверенных копий (например, фотокопии, микрофильмы, микрофиши или другие точные воспроизведения оригинальных документов). При использовании средств компактного хранения данных, например, микрофильмирования или записи в электронном виде следует иметь считывающее оборудование и средства для выполнения копий на жестких носителях.
- Спецификации на сырье, промежуточные продукты (при необходимости), АФС, печатные и упаковочные материалы должны быть оформлены в письменном виде. Могут потребоваться спецификации для других материалов (технологических добавок, прокладок и т. д.), используемых при производстве промежуточных продуктов или АФС, оказывающих решающее влияние на качество продукции. Критерии приемлемости для внутрипроизводственного контроля также должны быть оформлены в письменном виде.
- Электронные подписи на документах должны быть идентифицированы и защищены.
6.2 Протокол очистки и использования оборудования
-
- В протоколах использования, очистки, дезинфекции и/или стерилизации и обслуживания основного оборудования должны быть указаны дата, время, наименование и номер каждой серии произведенной на нем продукции, а также информация о лице, проводившем очистку и обслуживание этого оборудования.
-
- Не требуется составление отдельных протоколов очистки и использования оборудования в случае, если оно специально предназначено для производства одного промежуточного продукта или АФС и серии этого промежуточного продукта или АФС производятся в прослеживаемой последовательности. При использовании оборудования специального назначения протоколы очистки, обслуживания и использования могут быть частью протокола на серию продукции или быть оформлены в виде отдельных документов.
6.3 Протоколы на сырье, промежуточные продукты, печатные и упаковочные материалы для АФС
-
- Протоколы на сырье, промежуточные продукты, печатные и упаковочные материалы, используемые для производства АФС, должны храниться в установленном порядке и содержать следующие данные:
-
- наименование производителя, содержание и количество каждой поставки каждой партии сырья, промежуточных продуктов или печатных и упаковочных материалов для производства АФС, наименование поставщика, шифр(ы) или другой идентификационный номер поставщика (если известны), номер, присвоенный при поступлении, и дата поступления;
- результаты проведенных испытаний или исследований и выводы по ним;
- протоколы, прослеживающие использование материалов;
- протокол о соответствии материалов, используемых для маркировки и упаковки АФС, требованиям, установленным в спецификациях, и
- принятое решение относительно отклоненного сырья, промежуточных продуктов или печатных и упаковочных материалов, используемых в производстве АФС.
-
- Следует хранить утвержденные образцы этикеток для сравнения с ними выпускаемых этикеток.
6.4 Промышленные регламенты
-
- С целью обеспечения однообразия и повторяемости от серии к серии для каждого промежуточного продукта и АФС должны быть разработаны промышленные регламенты, подписанные одним лицом с указанием даты, проверенные и подписанные другим независимым лицом со стороны отдела(ов) качества с указанием даты.
-
- Промышленные регламенты должны содержать следующие данные:
- наименование производимого промежуточного продукта или АФС и ссылку на соответствующий документ (если это возможно);
- полный перечень сырья и промежуточных продуктов согласно их наименованиям или кодам, достаточным для идентификации всех их показателей качества;
- количество или соотношение используемых видов сырья или промежуточного продукта, в т. ч. единицу его измерения. Если это количество не является фиксированной величиной, то в протокол должен быть включен расчет для каждого размера серии или производительности. При наличии обоснования в протокол следует включать допустимые отклонения параметров;
- место нахождения производства и основное оборудование;
- подробные технологические инструкции, включающие в себя:
- последовательность операций;
- допустимые пределы изменения технологических параметров;
- инструкции по отбору проб и проведению внутрипроизводственного контроля с указанием критериев приемлемости (при необходимости);
- ограничения по времени завершения отдельных технологических стадий и/или всего технологического процесса;
- предполагаемый выход продукта для соответствующих стадий технологического процесса или отрезков времени;
- особые указания и меры предосторожности или ссылки на них (при необходимости);
- инструкции по хранению промежуточного продукта или АФС, печатных и упаковочных материалов и особых условий хранения с указанием сроков годности.
6.5 Протоколы на серию продукции (протоколы производства и контроля качества серии продукции)
-
- Для каждого промежуточного продукта и АФС составляют протоколы на серию продукции, включающие в себя полную информацию о производстве и контроле качества каждой серии. До выпуска продукции протокол на серию должен быть проверен с целью подтверждения содержащихся в нем данных и их соответствия промышленному регламенту. Если протокол на серию продукции относится к части промышленного регламента, в протоколе должна быть ссылка на соответствующий промышленный регламент.
-
- Протоколы на серию продукции должны быть пронумерованы с использованием индивидуального номера серии или идентификационного кода и подписаны с указанием даты. При непрерывном производстве до присвоения окончательного номера серии в качестве идентификационного кода допускается использовать шифр продукта с указанием даты и времени.
- В протоколах на серию продукции после завершения каждой важной технологической стадии следует указывать:
- дату и время, при необходимости;
- основное используемое оборудование (например, реакторы, сушильные аппараты, мельницы и т. д.);
- данные о каждой серии (в т. ч. взвешивание, отмеривание и номера серий сырья, промежуточных продуктов или любых других материалов, использованных в производстве);
- фактические данные критических технологических параметров;
- данные о всех отобранных пробах;
- подписи непосредственных исполнителей и лиц, проверяющих или контролирующих каждую критическую операцию в данном процессе;
- результаты внутрипроизводственного и лабораторного контроля;
- фактический выход продукции на соответствующих стадиях или на определенный момент времени;
- данные об упаковке и маркировке промежуточного продукта или АФС;
- образцы маркировки АФС или промежуточных продуктов, если их поставляют в готовом виде;
- любые отклонения, их оценку, результаты расследования (при необходимости) или ссылку на проведенное расследование (если оно хранится в виде отдельного протокола);
- результаты контроля при выпуске продукции.
-
- Следует разработать инструкции по расследованию критических отклонений и несоответствия серии промежуточных материалов или АФС требованиям спецификаций. В это расследование следует включать и другие серии продукции, которые могут быть связаны с данным несоответствием или отклонением.
6.6 Протоколы лабораторного контроля
-
- В протоколах лабораторного контроля должна быть полная информация о ходе всех испытаний, проведенных для подтверждения соответствия требованиям спецификаций и нормативной документации, в т. ч. данные наблюдений и анализов:
- описание полученных проб, в т. ч. наименование материала или место отбора пробы, номер серии или другой отличительный код, дата взятия образца, количество и дата поступления этой пробы в лабораторию;
- указание и/или ссылка на каждый использованный метод контроля;
- указание массы или размеров пробы для каждого испытания согласно принятой методике; данные или ссылка на приготовление и проведение испытания образцов сравнения, реактивов и стандартных растворов;
- необработанные в процессе каждого испытания данные в дополнение к графикам, таблицам и спектрам, полученным с использованием лабораторного оборудования с нумерацией (кодом), позволяющей определить испытуемое вещество и серию;
- вычисления с указанием единиц измерения, коэффициентов перевода и коэффициентов эквивалентности;
- результаты контроля и их сравнения с установленными критериями приемлемости;
- подпись ответственного лица, проводившего каждый вид контроля, и дату(ы) его проведения и
- дата и подпись второго лица, заверяющая точность, полноту и соответствие установленным требованиям.
-
- Следует также составлять протоколы с указанием:
- любых изменений в принятых аналитических методах;
- периодической калибровки (поверки) лабораторных приборов, оборудования и записывающих устройств;
- испытаний на стабильность АФС;
- результатов исследований, не удовлетворяющих требованиям спецификации.
6.7 Рассмотрение протоколов на серию продукции
-
- Для определения соответствия промежуточного продукта или АФС установленным требованиям до выпуска или реализации данной серии продукции следует разработать инструкции по рассмотрению и утверждению (согласованию) протоколов на серию продукции и проведению лабораторного контроля качества продукции, в т. ч. упаковку и маркировку.
- Протоколы на серию и лабораторные протоколы контроля критических технологических стадий до выпуска и реализации серии АФС должны быть рассмотрены и утверждены отделом(ами) контроля качества. Протоколы внутрипроизводственного и лабораторного контроля некритических технологических стадий могут быть рассмотрены квалифицированным производственным персоналом или другими отделами в соответствии с инструкциями, согласованными с отделом(ами) качества.
-
- Все отклонения от спецификаций и расследования этих случаев должны быть рассмотрены до выпуска серии в реализацию.
- Служба (отделы) контроля качества может передать ответственность и право выдачи разрешения на выпуск промежуточных продуктов производственному отделу (службе), за исключением случаев, когда промежуточные продукты поставляют за пределы зоны их контроля предприятиемпроизводителем.
7. Работа с материалами
7.1 Общий контроль
-
- Порядок получения, идентификации, карантинного хранения, обращения, отбора проб, проведения контроля и выдачи разрешения на использование или отклонение материалов должен быть установлен в соответствующих инструкциях.
- У производителей промежуточных продуктов и/или АФС должна быть разработана и внедрена система оценки поставщиков критических материалов.
- Поставка материалов должна осуществляться поставщиками, утвержденными отделом(ами) качества, в соответствии с согласованными спецификациями.
- Если поставщик критического материала не является производителем данного материала, производитель промежуточных продуктов и/или АФС должен иметь информацию о наименовании и адресе этого производителя.
- Замена поставщика критического сырья должна проводиться в соответствии с процедурой, изложенной в разделе 13.
7.2 Приемка и карантин
-
- Перед приемкой и при получении материалов на каждой упаковке или группе упаковок должны быть визуально проверены правильность маркировки (в т. ч. соответствие названия, используемого поставщиком и заказчиком, если они отличаются), сохранность упаковок, наличие поврежденных печатей, следов вскрытия и загрязнения упаковок. Материалы должны находиться на карантинном хранении до проведения отбора проб, их анализа (контроля) и получения разрешения на их использование.
- До смешивания поступивших материалов с имеющимися запасами (например, с растворителями или запасами, находящимися в хранилищах) следует установить их подлинность, при необходимости – провести контроль и получить разрешение на их использование. Для предотвращения неправильного размещения поступивших материалов среди уже хранящихся материалов должны быть разработаны специальные инструкции.
- Следует исключить возможность перекрестного загрязнения нерасфасованной продукции, если она поставляется в емкостях, не предназначенных специально для нее. В качестве доказательства этого могут использоваться:
- паспорт очистки;
- контроль на наличие следов примесей;
- аудит поставщика.
-
- Большие емкости для хранения и соответствующие коллекторы, линии по загрузке и выгрузке должны иметь соответствующую маркировку.
- Каждая емкость или группа емкостей (серий) материалов должна иметь четкую маркировку (шифр, номер серии или поставки, контракта, товарной накладной). Этот номер должен использоваться при обозначении расположения каждой серии. Должна быть разработана и внедрена система обозначения статуса каждой серии.
7.3 Отбор проб и проведение испытаний поступивших материалов
-
- Для подтверждения подлинности каждой серии материалов (за исключением материалов, указанных в 7.32) следует провести хотя бы одно испытание. Вместо проведения последующих испытаний можно использовать аналитический паспорт поставщика при условии, что на предприятии производителя действует система оценки поставщиков.
- Система оценки поставщиков должна включать в себя доказательства (например, данные о качестве предыдущих поставок) того, что производитель может стабильно поставлять материалы, соответствующие требованиям спецификации. Прежде чем упростить процедуру собственных проверок, следует провести полный анализ не менее трех серий материала. Полный анализ должен проводиться через определенные интервалы времени и сравниваться с аналитическим паспортом. Достоверность аналитического паспорта необходимо регулярно проверять.
-
- Не требуется проведение контроля технологических добавок, опасного или высокотоксичного сырья, других специальных материалов или материалов, передаваемых в другое подразделение под контролем заказчика, при наличии аналитического паспорта производителя, подтверждающего соответствие этого сырья установленным требованиям. Идентификацию этих материалов проводят путем визуального осмотра упаковок, этикеток и регистраций номеров серий. Если контроль таких материалов на месте не проводится, то это должно быть обосновано и оформлено документально.
- Пробы должны быть репрезентативной выборкой серии материала, из которой они отобраны. В методике отбора проб следует указывать число контейнеров, из которых должны быть взяты пробы, из какой части контейнера и в каком количестве их следует отбирать. Число контейнеров, из которых следует отобрать пробы, и количество проб должны быть установлены в плане отбора проб. Следует учитывать критичность и стабильность свойств материала, информацию о качестве предыдущих поставок и количестве материала, необходимого для проведения анализа.
- Отбор проб следует проводить в специально отведенном месте. Методика отбора проб должна предусматривать предотвращение загрязнения материала, из которого отбираются пробы, и других материалов.
- Упаковки, из которых отбирают пробы, должны быть аккуратно вскрыты и закрыты. На упаковках должна быть нанесена маркировка, показывающая, что из этой упаковки были взяты пробы.
7.4 Хранение
-
- При обращении с материалами и их хранении не допускаются ухудшение их качества, загрязнение и перекрестное загрязнение.
- Материалы, хранящиеся в картонных барабанах, мешках или коробках, должны быть расположены таким образом, чтобы было удобно проводить уборку и проверку. Не допускается складирование материалов на полу.
- Период и условия хранения материалов не должны отрицательно влиять на их качество. Как правило, материалы, поступившие первыми, следует использовать в первую очередь.
- Некоторые материалы можно хранить вне помещений при условии надлежащей упаковки, читаемой маркировки и выполнения необходимой очистки упаковок перед их вскрытием и использованием.
- Отклоненные материалы должны иметь соответствующую маркировку и помещаться на карантин (в изолятор брака) для исключения их несанкционированного использования в производстве.
7.5 Повторный контроль
7.50 При необходимости для определения пригодности материалов (например, после длительного хранения или в результате воздействия температуры или влажности) следует проводить повторный контроль.
8. Технологический процесс и внутрипроизводственный контроль
8.1 Технологические операции
-
- Условия взвешивания и отмеривания сырья для производства промежуточных продуктов и АФС не должны оказывать влияния на их пригодность. Устройства для взвешивания и отмеривания должны быть необходимой точности.
- Если материал разделяется на части для последующего использования в производственных операциях, он должен помещаться в специальную упаковку, маркировка которой содержит следующую информацию:
- наименование материала и/или код;
- номер, присвоенный при получении, или контрольный номер;
- массу или меру материала в новой упаковке и
- дату повторного контроля, при необходимости.
-
- Выполнение критических операций по взвешиванию, отмериванию или разделению материала на порции должно быть заверено или проконтролировано эквивалентным образом. До начала работы производственный персонал должен убедиться в том, что все полученные материалы соответствуют данным, указанным в протоколе серии для производства данного промежуточного продукта или АФС.
- Выполнение других критических действий должно быть заверено или проконтролировано эквивалентным образом.
- На определенных стадиях технологического процесса необходимо сравнивать фактический выход продукции (материалов) с ожидаемым. Ожидаемый выход и допустимые отклонения должны
быть определены на основании результатов лабораторных, опытных или производственных испытаний, проведенных предварительно. Отклонения от ожидаемого выхода, связанные с критическими технологическими стадиями, должны быть расследованы для определения их возможного влияния на качество серии продукции.
-
- Все отклонения должны быть оформлены документально с указанием причин. Любое критическое отклонение должно быть расследовано.
- Статус основных единиц оборудования в технологическом процессе должен быть указан на самом оборудовании или отражен в соответствующей документации, системе с компьютерным управлением и контролем или другим образом.
- Следует организовать контроль материалов, предназначенных для повторного использования или переработки во избежание их несанкционированного использования.
8.2 Ограничения на время выполнения операций
-
- Технологической инструкцией могут быть установлены ограничения на время выполнения операций (6.41). Отклонения от этих ограничений следует оформлять документально и проводить их оценку. Такие ограничения могут не потребоваться при проведении технологического процесса до достижения установленных значений параметров (например, доводка рН, гидрогенизация, сушка до значения, установленного спецификацией), так как в этом случае завершение реакций или стадий производства определяется при внутрипроизводственном отборе проб и контроле.
- Условия хранения промежуточных продуктов должны обеспечивать их дальнейшую пригодность.
8.3 Внутрипроизводственный отбор проб и контроль
-
- Для контроля и выполнения технологических стадий, которые влияют на показатели качества промежуточных продуктов и АФС, должны быть разработаны инструкции. Порядок проведения внутрипроизводственного контроля и критерии приемлемости определяют на основании информации, полученной при разработке технологического процесса или по предыдущему опыту производства.
- Критерии приемлемости, вид и объем испытаний могут зависеть от вида производимых промежуточных продуктов или АФС, проводимой реакции или технологической стадии и степени влияния этого процесса на качество продукции. На ранних технологических стадиях допускается проводить менее строгий внутрипроизводственный контроль; на более поздних технологических стадиях (например, на стадиях выделения и очистки) – контроль может быть более жестким.
- Порядок проведения внутрипроизводственного контроля на критических стадиях должен включать в себя контрольные точки и методы контроля, быть оформлен в письменном виде и согласован с отделом контроля качества.
- Внутрипроизводственный контроль может проводить персонал производственного подразделения, имеющий необходимую квалификацию, а наладку технологического процесса допускается проводить без разрешения службы контроля качества, если она осуществляется в установленных допустимых пределах, согласованных с отделом контроля качества. Результаты такого контроля должны быть документально оформлены в полном объеме и входить в протокол на серию продукции.
- Порядок отбора проб промежуточных материалов, продуктов и АФС должен быть приведен в документально оформленных и утвержденных в установленном порядке методиках. Планы и методики отбора проб должны быть обоснованными.
- Внутрипроизводственный отбор проб должен проводиться с использованием методик, предусматривающих предотвращение загрязнения материалов и других промежуточных продуктов или АФС. Применяемые методы должны обеспечивать целостность проб после их отбора.
- Если при проверке и/или наладке технологического процесса результаты внутрипроизводственного контроля окажутся не соответствующими требованиям спецификаций, то проведение анализа причин отклонений, как правило, не требуется.
8.4 Смешанные серии промежуточных продуктов или АФС
-
- В данном руководстве смешивание материалов определяется как процесс объединения материалов в рамках одной спецификации для производства однородного промежуточного продукта или АФС. Внутрипроизводственное смешивание фракций одной серии (например, объединение нескольких загрузок центрифуги из одной кристаллизационной серии или объединение фракций из нескольких серий для дальнейшего производства) рассматривается как часть технологического процесса и не считается смешиванием.
- Не допускается смешивание серии, не удовлетворяющей требованиям спецификации, с другими сериями. Каждая серия, вносимая в смесь, должна быть произведена согласно установленным требованиям, индивидуально испытана, и ее соответствие спецификации должно быть подтверждено до смешивания.
- Как правило, смешиваются:
- малые серии для увеличения ее размера;
- остатки (т.е. относительно малые количества выделенного материала) из серий одного и того же промежуточного продукта или АФС для формирования одной серии.
При необходимости допускаются и другие случаи смешивания материалов.
-
- Процессы смешивания следует контролировать и оформлять в документальном виде, а смешанная серия должна быть, при необходимости, испытана на соответствие установленным требованиям.
- В протоколе на серию, полученную после смешивания, должен прослеживаться процесс производства индивидуальных серий, формирующих эту смешанную серию.
- Если физические характеристики АФС являются критическими (например, АФС предназначена для использования в твердых дозированных формах для принятия внутрь или в суспензиях), то операции по смешиванию должны быть аттестованы для подтверждения однородности окончательной серии. Аттестация должна включать в себя испытание критических параметров (распределение частиц по размеру, объемная плотность при свободной засыпке и удельная плотность при уплотнении), на которые может повлиять процесс смешивания.
- Если смешивание материалов может оказать неблагоприятное воздействие на стабильность продукции, то следует провести испытания стабильности окончательной серии.
- Срок годности или дата проведения повторного контроля смешанной серии должны быть основаны на дате производства самой старой фракции или серии в смеси.
8.5 Контроль загрязнений
-
- Допускается переносить остатки материалов в последующие серии того же промежуточного продукта или АФС при наличии соответствующего контроля. К ним относятся остатки, прилипающие к стенкам измельчителя; слой влажных кристаллов, остающийся в центрифуге после разгрузки, и неполное удаление жидкостей или кристаллов из технологической емкости после передачи материала на следующую технологическую стадию. Такой перенос не должен приводить к переносу продуктов распада или микробного загрязнения, которые могут оказать отрицательное влияние на чистоту АФС.
- При выполнении технологических операций не следует допускать загрязнения промежуточных продуктов или АФС другими материалами.
- После проведения очистки оборудования следует принять меры предосторожности, чтобы избежать загрязнения АФС.
9. Упаковка и маркировка АФС и промежуточных продуктов
9.1 Общие положения
-
- Следует разработать и документально оформить методики на получение, идентификацию, карантинное хранение, отбор проб, проведение исследования и/или испытания и выдачу разрешения на использование, а также на обращение с упаковочными и печатными материалами.
- Материалы для упаковки и маркировки должны соответствовать установленным требованиям. Материалы, не соответствующие этим требованиям, должны отклоняться для предотвращения их использования в операциях, для которых они непригодны.
- Следует хранить протоколы на каждую поставку печатных и упаковочных материалов, содержащие информацию о поставке, осмотре или контроле этого материала, а также заключение о разрешении его использования или отклонения.
9.2 Упаковочные материалы
-
- Упаковочные материалы должны обеспечивать надлежащую защиту промежуточного продукта или АФС от порчи или загрязнения, которые могут произойти в процессе их транспортирования или хранения.
- Упаковочные материалы должны быть чистыми и, если это обусловлено видом промежуточного продукта или АФС, должны пройти обработку (дезинфекцию) для обеспечения их пригодности для дальнейшего использования. Упакованные материалы не должны вступать в химические реакции и обладать аддитивными или абсорбирующими свойствами, чтобы не допустить изменения качества промежуточного продукта или АФС за установленные пределы.
- При повторном использовании упаковки должны быть очищены в соответствии с утвержденными инструкциями, а все предыдущие этикетки удалены.
9.3 Выпуск и контроль печатных материалов
-
- Доступ к местам хранения печатных материалов должен быть разрешен только лицам, имеющим соответствующие полномочия.
- Порядок определения соответствия между количеством отпущенного, использованного и возвращенного печатного материала и оценки расхождений между количеством маркированных упаковок и выданных этикеток должен быть указан в инструкции. Факты несоответствия должны расследоваться, а результаты расследования должны быть утверждены отделом контроля качества.
- Весь неиспользованный печатный материал, содержащий номер серии или другую печатную информацию о серии, должен быть уничтожен. Возвращенный печатный материал должен храниться таким образом, чтобы предотвратить его перепутывание и обеспечить надлежащую идентификацию.
- Вышедший из употребления и устаревший печатный материал должен быть уничтожен.
- Следует организовать контроль за работой устройств, предназначенных для печатания этикеток для упаковочных операций, чтобы гарантировать соответствие всех оттисков протоколу на серию продукции.
- Следует организовать тщательный контроль этикеток, отпечатанных для серии, на их соответствие данной серии и установленным требованиям. Результат контроля должен быть оформлен документально.
- Образец используемой отпечатанной этикетки должен быть включен в протокол серии продукции.
9.4 Операции по упаковке и маркировке
-
- Следует разработать и документально оформить инструкции по использованию упаковочных и печатных материалов.
- Следует разработать инструкции по выполнению маркировки, исключающие перепутывание печатных материалов. Операции по маркировке различных промежуточных продуктов или АФС должны быть разграничены физически или пространственно.
- Этикетки, используемые на упаковках промежуточных продуктов или АФС, должны содержать наименование или идентификационный код, номер серии продукта и условия хранения (в том случае, когда эта информация является критической для обеспечения качества промежуточного продукта или АФС).
- Если промежуточный продукт или АФС предназначены для передачи за пределы зоны действия системы контроля их производителем, то маркировка также должна содержать наименование и адрес предприятия-производителя, количество содержимого, специальные условия транспортирования и любые другие требования, предусмотренные нормативной документацией. Если для промежуточных продуктов или АФС установлен срок годности, то он должен быть указан на маркировке и в аналитическом паспорте. Для промежуточных материалов или АФС, прошедших повторный контроль, дата повторного испытания должна быть указана на маркировке и в аналитическом паспорте.
- Упаковочное и печатное оборудование должно быть проверено непосредственно перед использованием для гарантии того, что все материалы, не нужные для последующей упаковки, были удалены. Эта проверка должна быть указана в протоколе на серию продукции, журнале оборудования или другой документации.
- В состав операций по упаковке входит проверка правильности маркировки первичных и вторичных упаковок промежуточных продуктов или АФС. Результаты этих проверок должны быть указаны в протоколах на серию продукции или контроль качества продукции.
- Промежуточные продукты или АФС, отправляемые за пределы зоны контроля производителя, должны быть опечатаны таким образом, чтобы при повреждении или отсутствии печати получатель обратил внимание на возможность замены содержимого.
10. Хранение и реализация
10.1 Хранение на складе
-
- В помещениях для хранения материалов должны быть предусмотрены соответствующие условия (например, поддерживание заданной температуры и влажности, при необходимости). Следует вести протоколы условий хранения, если они являются критическими для сохранения свойств материалов.
- Во избежание случайного или несанкционированного использования находящихся в карантине, отклоненных, возвращенных или отозванных материалов должны быть выделены отдельные зоны для их временного хранения до принятия решения об их дальнейшем использовании (если другое не указано в документации).
10.2 Реализация
-
- Реализация АФС и промежуточных продуктов третьим сторонам допускается только после получения разрешения отдела контроля качества на их выпуск. В условиях карантина АФС и промежуточные продукты могут быть переведены в другое подразделение при наличии разрешения отдела контроля качества, соответствующей документации и организации надлежащего контроля.
- Транспортирование АФС и промежуточных продуктов не должно оказывать влияния на их качество.
- Особые условия транспортирования и хранения АФС и промежуточных продуктов должны быть указаны на этикетке.
- Производитель должен убедиться в том, что подрядчик, осуществляющий транспортирование АФС и промежуточных продуктов по контракту, знает и соблюдает требуемые условия транспортирования и хранения.
- Для обеспечения возможности быстрого отзыва каждой серии продукции должна быть организована система отслеживания за ее реализацией.
11. Лабораторный контроль
11.1 Общий контроль
-
- Служба контроля качества должна иметь в своем распоряжении необходимые лабораторные помещения и оборудование.
- Следует разработать и документально оформить методики отбора проб, проведения испытаний, выдачи разрешения на использование или отклонение материалов, регистрации и хранения данных, полученных в лаборатории. Ведение протоколов и отчетов должно соответствовать требованиям 6.6.
- Все спецификации, планы отбора проб и методики проведения испытаний должны быть научно обоснованными и гарантировать, что сырье, АФС и промежуточные продукты, а также печатные и упаковочные материалы соответствуют установленным стандартам качества и/или чистоты. Спецификации и методики испытаний должны соответствовать требованиям, установленным при государственной регистрации. Допускается использовать другие спецификации в дополнение к входящим в регистрационное досье. Спецификации, планы отбора проб и методики испытаний, в т. ч. изменения к ним, должны быть разработаны соответствующим подразделением, рассмотрены и согласованы со службой (отделом) контроля качества.
- Спецификации для АФС должны быть разработаны в соответствии с действующими нормами и технологическим процессом, и включать в себя контроль за примесями (например, органическими и неорганическими примесями и остаточным содержанием растворителей). Если для АФС предусмотрены требования к микробиологической чистоте, то в спецификации должны быть установлены допустимые пределы для общего числа микроорганизмов и нежелательных микроорганизмов. Если для АФС предусмотрены требования к апирогенности, то в спецификации должны быть установлены допустимые пределы к уровню эндотоксинов.
- Все операции контроля в лабораториях должны проводиться в соответствии с утвержденными методиками (инструкциями) и оформляться в письменном виде непосредственно после их проведения. Любые отклонения от указанных методик должны быть обоснованы.
- Любые результаты, выходящие за рамки спецификаций, должны быть расследованы и оформлены в соответствии с принятой методикой, предусматривающей проведение анализа данных, оценку существенной проблемы, определение путей решения этой проблемы и формулирование выводов из проведенного расследования. После получения результатов, выходящих за рамки спецификаций, повторные отборы проб и/или повторные испытания должны проводиться в соответствии с принятыми методиками.
- Реактивы и стандартные растворы должны приготавливаться и маркироваться в соответствии с принятой методикой. Для аналитических реактивов и растворов следует применять маркировку «Использовать до».
- Для производства АФС необходимо иметь первичные стандартные образцы и документацию о происхождении каждого стандартного образца. Данные о хранении и использовании каждого первичного стандартного образца согласно рекомендациям поставщика должны быть приведены в соответствующих протоколах. Первичные стандартные образцы, полученные из официально признанных источников и хранящиеся в соответствии с рекомендациями поставщика, используют, как правило, без проведения испытаний.
- При невозможности получения первичных стандартных образцов из официально признанных источников следует разработать внутренние стандартные образцы производителя. Для установления подлинности и чистоты первичного стандартного образца требуется проведение соответствующего контроля. Проведение контроля должно быть оформлено документально.
- Следует разработать порядок изготовления, установления, идентификации, испытаний, утверждения и хранения вторичных стандартных образцов. Пригодность каждой серии вторичного стандартного образца должна быть определена до момента первого использования путем его сравнения с первичным стандартным образцом. Каждая серия вторичного стандартного образца должна периодически проходить проверку (аттестацию) в соответствии с утвержденной методикой.
11.2 Проведение испытаний промежуточных продуктов и АФС
-
- Для подтверждения соответствия каждой серии промежуточного продукта и АФС требованиям спецификаций должны быть проведены необходимые лабораторные испытания.
- Для каждой АФС, как правило, должен быть определен состав примесей (идентифицированных и неидентифицированных), присутствующих в обычной серии, произведенной в ходе конкретного контролируемого технологического процесса. Описание примесей должно включать в себя их идентификацию или какие-либо качественные аналитические параметры, позволяющие ее идентифицировать (например, время удерживания), диапазон содержания каждой выявленной примеси и их классификацию (неорганическая, органическая, растворитель). Состав примесей обычно зависит от технологического процесса и природы АФС. Для АФС, получаемых из растительного сырья или из тканей животных, определение состава примесей обычно не требуется. Требования к АФС, получаемым биотехнологическим путем, должны быть установлены в соответствующей нормативной документации.
- Для выявления изменений в АФС, происходящих вследствие изменений в сырье, оборудовании, рабочих параметрах или технологическом процессе, состав примесей периодически следует сравнивать с составом примесей, указанных в документации, оформляемой при государственной регистрации, или с данными, полученными ранее.
- Следует проводить контроль микробной чистоты каждой серии промежуточного продукта и АФС в соответствии с установленными требованиями.
11.3 Аттестация аналитических методик (см. раздел 12)
11.4 Аналитические паспорта
-
- На каждую серию промежуточного продукта и/или АФС должен выдаваться подлинный аналитический паспорт (по требованию).
- Аналитический паспорт должен содержать информацию о наименовании промежуточного продукта или АФС, в т. ч. о его чистоте, номере серии и дате выпуска. Для промежуточных продуктов и АФС с установленным сроком годности последний должен быть указан в маркировке и аналитическом паспорте. Для промежуточных продуктов или АФС с датой повторного контроля эта дата также должна быть указана в маркировке и/или аналитическом паспорте.
- В аналитическом паспорте должны быть перечислены все испытания, проведенные в соответствии с нормативными документами или требованиями заказчика, в т. ч. критерии приемлемости и полученные количественные результаты (при их наличии).
- В аналитическом паспорте должна быть указана дата и подпись ответственного лица отдела контроля качества, а также наименование, адрес и телефон первичного предприятияпроизводителя. Если анализ был проведен на вторичном предприятии-упаковщике или производителе, аналитический паспорт должен содержать наименование, адрес, телефон вторичного предприятия-упаковщика или производителя и наименование первичного производителя.
- Новые аналитические паспорта, выпускаемые вторичным предприятием-упаковщиком или производителем, поставщиками или лицами, действующими от их имени, должны содержать наименование, адрес и телефон лаборатории, проводившей эти анализы. В них также должны быть указаны наименование, адрес первичного производителя и оригинал паспорта на эту серию продукции с приложением его копии.
11.5 Контроль стабильности АФС
-
- Следует разработать и документально оформить программу непрерывного контроля стабильности АФС, результаты которого должны использоваться для подтверждения условий хранения, при пересмотре срока годности или срока проведения повторного контроля.
- Методики испытаний на стабильность должны быть аттестованы.
- Пробы для контроля стабильности следует хранить в упаковках, имитирующих коммерческие упаковки. Например, для АФС, поставляемых в мешках, помещенных в картонные барабаны, пробы для контроля стабильности могут быть упакованы в мешки из того же материала, помещенные в барабаны, меньшие по размеру и изготовленные из похожего или такого же материала, как и для АФС, поступающих в реализацию.
- Для подтверждения даты проведения повторного контроля или срока годности АФС первые три реализуемые серии, как правило, должны быть испытаны на стабильность. Если результаты предыдущих испытаний показывают, что данная серия АФС будет сохранять стабильность еще не менее двух лет, допускается использовать менее трех серий.
-
- В программу контроля стабильности ежегодно должна включаться, по крайней мере, хотя бы одна серия производимой АФС (за исключением случаев, когда в этот год ее производство не осуществлялось).
- Для АФС с коротким сроком годности испытания следует проводить чаще. Например, для биотехнологических/биологических и других АФС со сроком хранения один год и менее пробы на стабильность следует отбирать и испытывать ежемесячно в течение первых 3 мес. и затем с интервалом в 3 мес.
При наличии данных, подтверждающих сохранение стабильности АФС, может быть рассмотрен вопрос об увеличении периодичности испытаний (например, проведение испытаний через 9 мес.).
-
- Условия хранения при проведении испытаний на стабильность должны, по возможности, соответствовать требованиям нормативной документации к проведению этих испытаний (Руководство ICH).
11.6 Срок годности и дата повторного контроля
-
- Если промежуточный продукт предназначен для перемещения за пределы зоны действия системы обеспечения качества производителя и для этого продукта установлен срок годности или дата проведения повторного контроля, то следует иметь данные по его стабильности (например, опубликованную информацию, результаты испытаний).
- Срок годности АФС и дата повторного контроля должны быть основаны на результатах анализа данных, полученных при испытаниях на стабильность. Обычно для АФС используют дату повторного контроля, а не дату срока годности.
- Определение предварительного срока годности или даты повторного контроля АФС может основываться на данных производства опытных серий в следующих случаях:
- при производстве опытных серий использовались методы, моделирующие технологический процесс при промышленном производстве;
- качество АФС соответствует качеству продукции, которая будет производиться в промышленном масштабе.
-
- Для проведения повторного контроля должен использоваться образец, репрезентативный для этой серии.
11.7 Контрольные/архивные образцы
-
- Упаковка и хранение контрольных образцов предназначены для возможной оценки качества серий АФС в будущем, а не для проведения испытаний этих образцов на стабильность.
- Маркированные контрольные образцы из каждой серии АФС следует сохранять не менее одного года после истечения срока годности данной серии, определенного производителем, или в течение трех лет после даты реализации этой серии, в зависимости от того, какой срок больше. Для АФС с датой повторного контроля аналогичные контрольные образцы следует хранить в течение трех лет после того, как эта серия была полностью реализована производителем.
- Контрольные образцы должны храниться в такой же или в эквивалентной упаковке, используемой для АФС, либо в упаковке, более защищенной, чем упаковка, в которой реализуется эта продукция. Количество архивных образцов должно быть достаточным для проведения не менее двух полных анализов в соответствии с требованиями Фармакопеи, либо, в случае отсутствия соответствующей фармакопейной статьи, двух полных анализов согласно спецификации.
12. Аттестация (испытания)
12.1 Политика проведения аттестации (испытаний)
-
- Общая политика предприятия, цели и подход к процессам аттестации, в т. ч. к аттестации технологических процессов, методов очистки, аналитических методов, методов проведения внутрипроизводственного контроля, систем с компьютерным управлением и контролем и лиц, ответственных за разработку, рассмотрение, согласование и оформление каждой стадии аттестации (испытаний), должны быть оформлены в письменном виде.
- Как правило, в ходе разработки процесса (или по имеющимся данным) должны быть определены критические параметры (характеристики) и пределы их изменений, необходимые для обеспечения надежной работы. К таким параметрам относятся:
- критические характеристики, которые должны быть указаны для данного продукта (АФС);
- технологические параметры, которые могут повлиять на критические характеристики качества
АФС;
- предельные изменения каждого критического параметра процесса, которые будут использоваться в ходе текущего производственного контроля.
-
- Операции, которые считаются критическими для качества и чистоты АФС, подлежат аттестации.
12.2 Документация по аттестации (испытаниям)
-
- Для каждого процесса, подлежащего аттестации, следует разработать методику проведения аттестации, согласованную между службой качества и другими отделами, имеющими к ней отношение.
- В методике аттестации должны быть определены критические операции (стадии) технологического процесса и критерии приемлемости, а также виды аттестации (например, ретроспективная, перспективная, текущая) и количество производственных циклов, необходимых для проведения аттестации.
- Отчет о проведении аттестации должен содержать ссылку на методику аттестации (с указанием всех замеченных отклонений) и выводы с рекомендациями по устранению недостатков.
- Любые отклонения от требований методики аттестации должны быть обоснованы и оформлены в письменном виде.
12.3 Этапы аттестации (испытаний)
12.30 До проведения работ по аттестации процессов следует провести аттестацию критического оборудования и вспомогательных систем в необходимом объеме. Как правило, такую аттестацию проводят в несколько этапов:
- аттестация проекта (Design Qualification; DQ) – документальное подтверждение того, что проект производства (в т. ч. помещения, оборудование или вспомогательные системы) соответствует своему назначению*;
- аттестация установленного оборудования (Installation Qualification; IQ) – документальное подтверждение того, что установленное или измененное оборудование, помещения или вспомогательные системы соответствуют утвержденному проекту, заданию на проектирование и действующим нормам;
- аттестация функционирующего оборудования (Operational Qualification; OQ) – документальное подтверждение того, что установленное или измененное оборудование, помещения или вспомогательные системы функционируют надлежащим образом в требуемом режиме;
- аттестация в эксплуатации (Performance Qualification; PQ) – документальное подтверждение того, что оборудование, помещения и вспомогательные системы могут работать совместно с нужной эффективностью и воспроизводимостью в соответствии с утвержденными требованиями и характеристиками технологического процесса.
12.4 Аттестация (испытания) процесса
-
- Аттестация (испытания) процесса — документальное подтверждение того, что процесс, выполняемый в пределах установленных параметров, обеспечивает эффективное и воспроизводимое производство промежуточных продуктов и АФС, удовлетворяющих установленным требованиям и показателям качества.
- Существуют три вида аттестации. Из трех видов аттестации предпочтительным является перспективная аттестация, но возможно использование и других видов. Эти виды аттестации и области их применения приводятся ниже.
- Перспективную аттестацию применяют для всех процессов производства АФС, определенных в 12.12. Перспективная аттестация, используемая для процесса производства АФС, должна быть завершена до выпуска на рынок готового лекарственного средства, произведенного из этой АФС.
- Текущая аттестация может проводиться при отсутствии данных о повторных технологических циклах. Это может быть обусловлено производством только ограниченного числа серий АФС, нерегулярным производством серий АФС или производством АФС с использованием аттестованного процесса, в который были внесены изменения. До завершения текущей аттестации серии АФС могут быть выпущены и использованы для производства готовых лекарственных средств для их реализации на рынке на основании тщательного непрерывного контроля и проведения испытаний этих серий АФС.
- Ретроспективную аттестацию применяют как исключение для хорошо отлаженных процессов, которые не претерпевали значительных изменений для качества АФС из-за изменений сырья, оборудования, систем, устройств или применяемого технологического процесса. Этот вид аттестации
* Заданию на проектирование и действующим нормам (прим. разработчика стандарта).
может использоваться в том случае, если:
- определены критические характеристики качества и критические параметры процесса;
- установлены соответствующие внутрипроизводственные критерии приемлемости и контроля;
- отсутствовали существенные сбои в ходе процесса (качестве продукции) по причинам, не связанным с ошибками оператора или отказами оборудования;
- установлен состав примесей для существующих АФС.
-
- Серии продукции, отобранные для проведения ретроспективной аттестации, должны представлять собой репрезентативную выборку из всех серий, произведенных за рассматриваемый период времени, в т. ч. всех серий, которые не соответствовали требованиям спецификаций. Число этих серий должно быть достаточным для подтверждения стабильности процесса. При проведении ретроспективной аттестации допускается использовать контрольные (архивные) образцы.
12.5 Программа аттестации (испытаний) процесса
-
- Число производственных циклов для проведения аттестации (испытаний) зависит от сложности процесса или характера изменения параметров процесса. Для проведения перспективной и текущей аттестации следует использовать не менее трех последовательно произведенных серии продукции надлежащего качества. Однако могут быть случаи, при которых для доказательства стабильности процесса могут потребоваться дополнительные производственные циклы (например, сложные или длительные процессы производства АФС). При ретроспективных испытаниях для подтверждения стабильности процесса исследуют данные 10 – 30 последовательных серий продукции. При наличии обоснования это число может быть уменьшено.
- В ходе аттестации (испытаний) процесса следует контролировать его критические параметры. Параметры процесса, не связанные с качеством продукции (например, расход энергии или срок использования оборудования), не подлежат аттестации.
- Аттестация процесса должна подтвердить, что установленный состав примесей АФС находится в допустимых пределах. Состав примесей (качественный и количественный) должен быть аналогичен или лучше состава примесей, установленного по предыдущим данным, или, где применимо, быть лучше состава примесей, установленного при разработке технологического процесса; или профиля примесей в сериях продукции, предназначенных для проведения клинических или токсикологических исследований.
12.6 Периодическая оценка аттестованных (испытанных) систем и процессов
12.60 Системы и процессы следует периодически оценивать для подтверждения того, что они продолжают работать в аттестованном режиме. Повторная аттестация не требуется, если в систему или процесс не было внесено никаких существенных изменений и анализ качества продукции подтверждает, что эта система или процесс стабильно производят продукцию в соответствии с установленными требованиями.
12.7 Аттестация методов очистки
-
- Как правило, следует проводить аттестацию методов очистки. Обычно аттестацию методов очистки проводят в случаях, когда загрязнение или перенос материалов представляют наибольший риск для качества АФС. Например, на ранних стадиях производства может оказаться ненужной аттестация методов очистки оборудования, если все остатки удаляются последующими операциями очистки.
- Аттестация методов очистки должна отражать фактический характер использования оборудования. Если на одном и том же оборудовании производятся различные АФС или промежуточные продукты, и очистка этого оборудования проводится одним и тем же способом, то для аттестации очистки может быть выбран репрезентативный образец промежуточного продукта или АФС. Этот выбор должен быть основан на свойствах растворимости вещества и трудности его очистки, а также на расчете допустимых предельных значений остатков на основе его активности, токсичности и стабильности.
- Методика аттестации очистки должна содержать характеристику оборудования, подлежащего очистке, порядок очистки, перечень материалов, допустимые предельные значения чистоты, параметры, которые следует контролировать, и применяемые аналитические методы. В методике также следует указать вид отбираемых проб, методы их отбора и маркировки.
- Для определения растворимых и нерастворимых остатков при отборе проб следует использовать такие методы, как взятие мазков, смывов, или альтернативные методы (например, прямую экстракцию). Используемые методы отбора проб должны позволять проводить количественное определение остатков на поверхности оборудования после его очистки. Отбор проб путем взятия мазков может быть не всегда применим, если поверхности контакта с продуктом являются труднодоступными из-за особенностей конструкции оборудования и/или ограничений процесса (например,внутренние поверхности шлангов, труб, сосудов, реакторов с маленькими отверстиями) или при работе с токсическими материалами, а также с малогабаритным сложным оборудованием (например, с измельчителями и псевдоожижителями).
-
- Следует использовать аттестованные аналитические методы, обладающие достаточной чувствительностью для определения допустимого уровня остатков или загрязнений. Для каждого метода следует определить его воспроизводимость. Предельные допустимые количества остатков должны быть реально достижимыми, проверяемыми и основываться на остатке наиболее опасного вещества. Предельные допустимые уровни остатков могут устанавливаться на основе минимальной известной фармакологической, токсикологической или физиологической активности АФС или ее наиболее опасного компонента.
- Для процессов, в которых существует ограничение на общее микробное загрязнение или уровень эндотоксинов в АФС, или других процессов, для которых такое загрязнение может представлять опасность (например, при производстве нестерильных АФС, используемых в производстве стерильных препаратов), аттестация очистки (дезинфекции) оборудования должна включать в себя определение уровня загрязнения по микроорганизмам и эндотоксинам.
- После проведения аттестации методы очистки должны контролироваться с определенной периодичностью для подтверждения того, что они сохраняют эффективность при использовании в текущем производстве. Чистоту оборудования можно контролировать аналитическими методами, а если это допускается, – путем визуального осмотра. Визуальный осмотр позволяет определить большие скопления загрязнений на малых площадях, которые могут оставаться необнаруженными при отборе проб и/или проведении анализов.
12.8 Аттестация аналитических методов
-
- Следует предусматривать аттестацию аналитических методов, за исключением фармакопейных или других утвержденных методов. В каждом конкретном случае следует подтвердить и оформить документально пригодность каждого применяемого аналитического метода.
- При аттестации аналитических методов следует проверять характеристики, включенные в Руководство ICH по аттестации аналитических методов. Объем исследований, выполняемых при аттестации аналитических методов, должен соответствовать целям анализа и стадии процесса производства АФС.
- Перед началом аттестации аналитических методов следует провести аттестацию соответствующего аналитического оборудования.
- Протоколы всех изменений, вносимых в аттестованный аналитический метод, следует сохранять. В протоколах должны быть указаны причина, из-за которой было внесено изменение, и данные, подтверждающие, что точность и достоверность результатов, полученных после внесения этого изменения, соответствуют характеристикам аттестованного метода.
13. Контроль изменений
-
- На предприятии должна быть введена и документально оформлена система контроля изменений, способных влиять на производство и качество промежуточных продуктов и АФС.
- Документальное оформление, рассмотрение и утверждение изменений в сырье, спецификациях, аналитических методах, помещениях, вспомогательных системах, оборудовании (в т. ч. в компьютерном оборудовании), технологических стадиях, печатных и упаковочных материалах и компьютерных программах должны выполняться по утвержденным инструкциям.
- Все предложения по изменениям, касающимся соблюдения требований настоящего стандарта, должны быть подготовлены, рассмотрены и согласованы с соответствующими подразделениями и службой качества.
- Следует оценить возможное влияние предложенных изменений на качество промежуточных продуктов и АФС. При этом может проводиться классификация изменений для определения объема работ при аттестации. Изменения могут быть классифицированы (например, как незначительные или существенные) в зависимости от их характера и степени влияния, которые эти изменения могут оказать на данный процесс. Следует определить и обосновать, какие дополнительные испытания и аттестационные исследования необходимы для подтверждения возможности внесения изменений в аттестованный процесс.
- При внесении изменений следует принять меры по пересмотру всех документов, на содержание которых влияют эти изменения.
- После внесения изменений следует оценить качество первых серий продукции, произведенных или испытанных с учетом этих изменений.
- Следует оценить возможность влияния критических изменений на установленные сроки годности или дату повторного контроля. При необходимости образцы промежуточных продуктов или АФС, произведенных по модифицированному процессу, могут быть включены в программу ускоренного определения стабильности и/или в программу непрерывного контроля стабильности.
-
- Все производители готовых лекарственных средств, которым поставляют АФС, должны быть извещены об изменениях установленных методов производства и контроля качества, способных оказать влияние на качество АФС.
14. Отклонение и переработка материалов
14.1 Отклонение
14.10 Следует выполнить маркировку промежуточных продуктов и АФС, не удовлетворяющих требованиям спецификаций, и поместить их на карантинное хранение. Эти промежуточные продукты и АФС могут быть вновь использованы в производстве или направлены на переработку в приведенном ниже порядке. Окончательное решение об использовании (утилизации) отклоненных материалов должно быть оформлено документально.
14.2 Повторная обработка
-
- Как правило, допускается повторное использование промежуточных продуктов и АФС (в т. ч. материалов, не удовлетворяющих требованиям спецификации) в производстве и их повторная обработка путем повторения стадии кристаллизации или других физических или химических стадий обработки (например, дистилляция, фильтрация, хроматография, измельчение), входящих в технологический процесс. Однако в том случае, если такие операции проводят при производстве большинства серий продукции, они должны быть включены в производство в качестве отдельной стадии принятого технологического процесса.
- Продолжение технологической стадии после того, как внутрипроизводственный контроль показал, что эта стадия еще не завершена, рассматривают как часть процесса и не считают повторной обработкой.
- Введение не вступившего в реакцию материала вновь в процесс и повторение химической реакции считают повторной обработкой, если такая операция не является частью технологического процесса. Такой повторной обработке должна предшествовать тщательная оценка, гарантирующая, что качество промежуточных продуктов и АФС не ухудшится вследствие возможного образования побочных продуктов и веществ при дальнейшем протекании реакции.
14.3 Переработка
-
- Перед принятием решения о переработке серии продукции, не соответствующей установленным требованиям, следует провести расследование причин такого несоответствия.
- Для серий продукции, полученных в результате переработки, следует провести ее оценку, испытания и, при необходимости, — испытания на стабильность с документальным оформлением для подтверждения соответствия ее качества качеству продукции, произведенной по базовой технологии. Для методик переработки может использоваться подход текущей аттестации. Это позволяет отработать методику и определить ожидаемые результаты. Допускается единичный выпуск серии переработанной продукции на основании отчета о ее производстве, если установлена такая возможность.
- Следует разработать методики сравнения состава примесей в каждой серии переработанной продукции с составом примесей в сериях продукции, произведенных по базовой технологии. Если для описания характеристик серии переработанной продукции недостаточно обычного набора аналитических методов, следует применять дополнительные методы.
14.4 Регенерация материалов и растворителей
-
- Допускается регенерация реактивов, промежуточных продуктов или АФС (например, из маточных растворов или фильтратов) при наличии утвержденных методик регенерации и соответствия качества регенерированных материалов установленным требованиям.
- Допускается регенерация и повторное использование растворителей в том или другом процессе при условии контроля процессов их регенерации и подтверждения соответствия растворителей установленным требованиям до их повторного использования или смешивания с другими материалами.
- Допускается смешивание свежеприготовленных и регенерированных растворителей и реактивов, если при испытаниях была установлена возможность их применения во всех технологических процессах, в которых они могут использоваться.
- Использование регенерированных растворителей, маточных растворов и других регенерированных материалов должно быть оформлено документально.
14.5 Возврат
-
- Возвращенные промежуточные продукты или АФС должны иметь соответствующую маркировку и помещаться на карантинное хранение.
- Если условия, в которых хранились или транспортировались возвращенные промежуточные продукты или АФС до или в процессе их возврата, или состояние их упаковок вызывают сомнения относительно качества этих материалов, то возвращенные промежуточные продукты или АФС должны быть переработаны, повторно обработаны или уничтожены в установленном порядке.
- Следует хранить протоколы о всех возвращенных промежуточных продуктах или АФС. Протокол каждого возврата должен содержать:
- наименование и адрес грузополучателя;
- наименование промежуточного продукта или АФС, номер серии и возвращенное количество;
- причину возврата;
- указание на использование или уничтожение возвращенного промежуточного продукта или АФС.
15. Рекламации и отзывы
-
- Все рекламации по качеству, полученные в устной или письменной форме, должны регистрироваться и расследоваться в соответствии с утвержденной инструкцией.
-
- Протоколы рассмотрения рекламаций должны включать в себя:
- имя, отчество и фамилию, а также адрес лица, предъявившего рекламацию;
- имя, отчество и фамилию, а также, должность и номер телефона лица, принявшего эту рекламацию;
- суть рекламации (с указанием наименования и номера серии АФС);
- дату поступления рекламации;
- первоначально принятые меры (с указанием ответственных лиц и даты);
- дальнейшие действия;
- ответ на рекламацию (с указанием даты отправки ответа);
- окончательное решение по серии или партии промежуточного материала или АФС.
-
- Протоколы рассмотрения рекламаций следует хранить для оценки тенденций, частоты и серьезности рекламаций по данному продукту и принятия дополнительных и срочных мер по устранению недостатков, при необходимости.
- Решение об отзыве следует принимать на основе инструкции, определяющей условия от-
зыва.
-
- В инструкции по отзыву продукции должны быть указаны лица, участвующие в анализе поступившей информации, процедуры начала отзыва продукции, круг лиц, которых следует проинформировать об отзыве, и порядок использования (утилизации) отозванного материала.
-
- В случае серьезной или потенциальной опасности для жизни населения следует проинформировать местные, национальные и/или международные компетентные органы и обратиться к ним с просьбой о содействии.
16. Работа по контракту (в т. ч. проведение анализов)
-
- Все производители, работающие по контракту (в т. ч. лаборатории), должны выполнять требования настоящего стандарта. Особое внимание должно быть уделено вопросам предотвращения перекрестного загрязнения и обеспечению прослеживаемости.
- Заказчик должен оценить способность подрядчика осуществлять на месте делегированные ему операции в соответствии с требованиями настоящего стандарта.
- Ответственность заказчика и подрядчика за соблюдение требований настоящего стандарта, в т. ч. по обеспечению качества, должна быть определена в контракте (договоре) между ними.
- Договор должен позволять заказчику проводить аудит производства подрядчика на его соответствие настоящему стандарту.
- Если допускается работать по договору субподряда, то подрядчик не должен передавать третьей стороне никакой части работы, порученной ему заказчиком, без предварительного согласования с ним.
- Производственные и лабораторные протоколы должны храниться в местах проведения этого вида деятельности и быть легкодоступными.
- Не допускается внесение изменений в технологический процесс, оборудование, спецификации, методы испытаний и др., относящиеся к предмету договора, без согласования с заказчиком.
17. Реализация, хранение, переупаковка и перемаркировка
17.1 Область применения
-
- Этот раздел относится к любым действиям (за исключением производства), связанным с реализацией, хранением, переупаковкой и перемаркировкой промежуточных продуктов или АФС.
- Все лица и организации, связанные с реализацией, хранением, переупаковкой и перемаркировкой промежуточных продуктов или АФС, должны соблюдать требования настоящего стандарта и, в частности, данного руководства.
17.2 Прослеживаемость реализуемых промежуточных продуктов или АФС
17.20 При реализации, хранении, переупаковке и перемаркировке необходимо обеспечить полную прослеживаемость реализуемых промежуточных продуктов или АФС. Для этого следует иметь и хранить следующие документы:
- реквизиты первичного предприятия-производителя;
- адрес первичного предприятия-производителя;
- заказы (договоры) на поставку;
- накладные (транспортные документы);
- документы о получении грузов;
- наименования или обозначения промежуточных продуктов или АФС;
- номер серии продукции, указанной производителем;
- документацию на транспортирование и реализацию;
- все подлинные аналитические паспорта, в т. ч. паспорта первичного предприятияпроизводителя;
- дату проведения повторного контроля или срок годности.
17.3 Обеспечение качества
17.30 Реализация, хранение, переупаковка и перемаркировка промежуточных продуктов или АФС должны проводиться в соответствии с документально оформленной системой обеспечения качеством (см. п. 2).
17.4 Переупаковка, перемаркировка и обращение с промежуточными продуктами или АФС
-
- Во избежание перепутывания и ухудшения качества и чистоты промежуточных продуктов или АФС переупаковка, перемаркировка и обращение с промежуточными продуктами или АФС и их контроль должны проводиться в соответствии с требованиями данного стандарта.
- Во избежание прямого и перекрестного загрязнения переупаковка продукции должна проводиться в установленных условиях окружающей среды.
17.5 Стабильность
17.50 Для подтверждения установленных сроков годности или даты повторного контроля исследования стабильности должны проводиться в случаях, если АФС или промежуточный продукт переупаковывают в упаковку другого типа, отличающуюся от используемой производителем.
17.6 Передача информации
-
- При реализации, хранении, переупаковке и перемаркировке промежуточных продуктов или АФС следует передавать заказчикам всю информацию, получаемую от производителя АФС или промежуточных продуктов, касающуюся качества продукции и решений уполномоченных органов, а также передавать производителям информацию от заказчиков.
- При реализации, хранении, переупаковке и перемаркировке промежуточных продуктов или АФС следует предоставлять потребителю наименование первичного предприятия-производителя АФС или промежуточных продуктов и номера поставляемых серий продукции.
- По требованию уполномоченного органа должна предоставляться информация о первичном предприятии-производителе промежуточных продуктов или АФС. Первичное предприятиепроизводитель может контактировать с уполномоченным органом непосредственно или через своих представителей в зависимости от взаимоотношений между этим предприятием и его представителем.
- Следует соблюдать специальные требования к аналитическим паспортам, приведенные в 11.4.
17.7 Работа с рекламациями и отзывами
-
- Все лица и организации, связанные с реализацией, хранением, переупаковкой и перемаркировкой промежуточных продуктов или АФС, должны хранить протоколы всех поступивших рекламаций и отзывов в соответствии с требованиями, указанными в разделе 15 настоящего руководства.
- При необходимости эти лица и организации, а также производитель АФС или промежуточного продукта должны рассмотреть рекламацию для принятия решения о дальнейших действиях, в которых могут участвовать другие потребители (заказчики), получавшие этот АФС или промежуточный продукт, уполномоченный орган или все заинтересованные стороны. Соответствующей стороной должно быть проведено расследование рекламации или отзыва продукции, результаты которого должны быть оформлены документально.
- Если рекламация касается производителя АФС или промежуточного продукта, то документация, предоставляемая агентами, брокерами, трейдерами, дистрибьюторами, лицами, выполняющими переупаковку и перемаркировку, должна содержать любые данные, полученные от данного производителя АФС или промежуточного продукта (в т. ч. дату и необходимую информацию).
17.8 Работа с возвратами
17.80 Работу с возвратом продукции проводят в соответствии с требованиями 14.52. Все лица и организации, связанные с реализацией, хранением, переупаковкой и перемаркировкой промежуточных продуктов или АФС, должны вести документацию по возвращенным АФС и промежуточным продуктам.
18. АФС, производимые путем культивирования клеток (ферментации)
18.1 Общие положения
-
- В настоящем разделе приведены специальные правила производства и контроля качества АФС или промежуточных продуктов путем культивирования (ферментации) клеток с использованием природных или рекомбинантных организмов, которые не были в должной мере указаны в предыдущих разделах. Этот раздел нельзя отделить от общего содержания настоящего стандарта. Все требования, установленные в настоящем стандарте, остаются в силе. Следует обратить внимание на то, что принципы ферментации для «классических» процессов получения низкомолекулярных соединений и процессов, использующих рекомбинантные и нерекомбинантные организмы для получения белков и/или полипептидов, одинаковы, хотя при этом уровень контроля этих процессов будет различным. В случае, если это имеет значение, в данном разделе указываются эти различия. В целом уровень контроля биотехнологических процессов, используемых для производства белков и полипептидов, должен быть более строгим, чем контроль над классическими процессами ферментации.
- Термин «биотехнологический процесс» относится к использованию клеток или организмов, которые для производства АФС были получены или модифицированы с использованием рекомбинантной ДНК, гибридомной или какой-либо другой технологии. АФС, произведенные биотехнологическими методами, обычно представляют собой высокомолекулярные соединения, такие как белки и полипептиды, для которых в этом разделе приведены специфические требования. С помощью технологии рекомбинантной ДНК могут быть также произведены некоторые АФС с низкими молекулярными массами, например, антибиотики, аминокислоты, витамины и углеводы. Уровень контроля за такими АФС должен соответствовать правилам, применяемым для классической ферментации.
- Термин «классическая ферментация» относится к процессам, использующим для производства АФС микроорганизмы, существующие в природе или модифицированные традиционными методами (радиацией, химическим мутагенезом). АФС, получаемые методом «классической ферментации», обычно являются низкомолекулярными соединениями, такими как антибиотики, аминокислоты, витамины и углеводы.
- Получение АФС и промежуточных продуктов из культуры клеток или методом ферментации включает в себя такие биологические процессы, как культивирование клеток или экстракция и очистка веществ из живых организмов. Следует обратить внимание на то, что могут существовать и дополнительные стадии (например, физико-химическая модификация), которые являются частью технологического процесса. Используемые исходные материалы (питательные среды, компоненты буфера) могут обеспечивать возможность роста микробных загрязнений. В зависимости от природы, метода приготовления и целей использования АФС или промежуточного продукта на определенных стадиях производства может потребоваться проведение контроля микробного, вирусного загрязнения и/или уровня эндотоксинов данной продукции.
- Для обеспечения качества АФС и промежуточных продуктов на всех стадиях производства должен быть установлен надлежащий контроль. Так как настоящий стандарт начинается со стадии культивирования клеток (ферментации), предыдущие стадии (например, поддерживание банка клеток) должны проводиться при наличии надлежащего внутрипроизводственного контроля. Настоящий
стандарт описывает процесс культивирования (ферментации) клеток с момента, когда из банка клеток поступает емкость с культурой клеток для использования в производстве.
-
- Для сведения риска загрязнения к минимуму следует использовать необходимое оборудование и проводить контроль окружающей среды. Критерии приемлемости качества и периодичность проведения контроля окружающей среды зависят от технологической стадии и условий производства (открытая, закрытая или замкнутая система).
- В целом, технологический контроль должен учитывать:
- поддерживание в рабочем состоянии рабочего банка клеток;
- правильную инокуляцию и развитие культуры клеток;
- контроль критических рабочих параметров во время ферментации/ культивирования клеток;
- непрерывный контроль процесса роста клеток, их жизнеспособности (для большинства процессов культивирования клеток) и продуктивности;
- методы сбора и очистки, при которых происходит удаление клеток, клеточного дебриса и компонентов питательной среды с одновременной защитой промежуточного продукта или АФС от загрязнения (в частности, микробиологического характера) и потери качества;
- непрерывный контроль микробного загрязнения и, при необходимости, уровня эндотоксинов на определенных стадиях производства, и
- вопросы защиты от вирусного загрязнения, описанные в Руководстве ICH Q5A*.
-
- При необходимости следует показать, что остатки среды, белки клетки-хозяина, другие примеси и загрязнения, связанных с технологическим процессом, удалены.
18.2 Поддерживание банков клеток и ведение протоколов
-
- Доступ к банкам клеток должен быть ограничен лицами, имеющими на это полномочия.
- Банки клеток должны храниться в специально разработанных условиях, обеспечивающих поддерживание жизнеспособности и предотвращающих загрязнение клеток.
- Следует вести и сохранять протоколы использования образцов из банков клеток и условия их хранения.
- Банки клеток должны периодически проходить контроль пригодности их использования.
- Более подробная информация по содержанию банков клеток изложена в Руководстве ICH Q5D**.
18.3 Культивирование клеток (ферментация)
-
- Закрытые системы следует применять, по возможности, в тех случаях, когда необходимо добавлять клеточные субстраты, среды, буфер и газы в асептических условиях. Если первичная инокуляция, последующий перенос или добавки (среды, буфер) проводятся в открытых сосудах, для снижения риска заражения должны быть разработаны и применяться соответствующие методы, а также проводиться контроль.
- Если микробное загрязнение может оказать влияние на качество АФС, операции с использованием открытых сосудов следует проводить в биобезопасных шкафах (боксах) или других устройствах, обеспечивающих контроль условий окружающей среды.
- При работе с культурами клеток персонал должен быть одет в специальную одежду и соблюдать специальные меры предосторожности.
- Следует проводить непрерывный контроль критических рабочих параметров (например, температуры, рН, скорости перемешивания, добавления газов, давления), который позволяет гарантировать соответствие условий установленным параметрам процесса. Следует также постоянно контролировать рост клеток, жизнеспособность (для большинства культивируемых клеток) и их продуктивность. В зависимости от типа процесса критические параметры могут изменяться, а в случае классической ферментации некоторые параметры можно не контролировать (например, жизнеспособность клеток).
- Оборудование, используемое для культивирования клеток, после окончания процесса должно быть очищено и стерилизовано. Оборудование для проведения ферментации также должно быть очищено, дезинфицировано или стерилизовано.
- Для предотвращения неблагоприятного влияния на качество АФС следует стерилизовать среды культивирования перед использованием.
- Методики обнаружения загрязнения и принятия соответствующих мер должны быть
- Руководство ICH Q5A «Качество биотехнологических продуктов. Оценка безопасности вирусного заражения биотехнологических продуктов, произведенных из клеток человеческого и животного происхождения» (Quality of Biotechnological Products: Viral Safety Evaluation of Biotechnology Products Derived from Cell Lines of Human or Animal Origin).
** Руководство ICH Q5D «Качество биотехнологических продуктов. Выделение и характеристика клеточных субстратов, используемых для производства биотехнологических/биологических продуктов» (Quality of Biotechnological Products: Derivation and Characterization of Cell Substrates Used for Production of Biotechnological/Biological Products).
оформлены документально. К ним относятся методики определения влияния загрязнения на продукцию, методы очистки оборудования и приведение оборудования в состояние готовности для дальнейшего использования. Посторонние микроорганизмы, обнаруженные в процессе ферментации, должны быть идентифицированы, а влияние их на качество продукции должно быть определено. Результаты таких оценок должны приниматься во внимание при решении вопроса об использовании произведенной продукции.
-
- Протоколы о случаях загрязнения следует сохранять.
- После очистки универсального оборудования между циклами производства различной продукции может потребоваться проведение дополнительных испытаний с целью снижения риска перекрестных загрязнений.
18.4 Сбор, выделение и очистка
-
- Операции по сбору материала (удалению клеток или клеточных компонентов или сбору клеточных компонентов после разрушения клеток) должны проводиться на оборудовании и в зонах, проект (конструкция) которых должен предусматривать снижение риска загрязнения до минимума.
- Следует разработать методики сбора и очистки, приводящие к удалению или инактивации организмов-продуцентов, клеточного дебриса и компонентов среды (оказывающих при этом минимальное влияние на деградацию, загрязнение и потерю качества) и гарантирующие выход промежуточного продукта и АФС надлежащего качества.
- После использования оборудование должно быть очищено и дезинфицировано в установленном порядке. Производство нескольких последовательных серий промежуточного продукта и АФС без промежуточной очистки оборудования допускается только в том случае, если это не оказывает влияния на их качество.
- При использовании открытых систем очистка должна проводиться в условиях, обеспечивающих сохранение качества продукции.
- Если оборудование используется для производства различных видов продукции, то могут применяться дополнительные виды контроля, такие как использование специальных хроматографических смол или проведение дополнительных испытаний.
18.5 Удаление вирусов (стадии инактивации)
-
- Более полная информация приведена в Руководстве ICH Q5A*.
- Стадии удаления и инактивации вирусов являются критическими для некоторых технологических процессов и должны проводиться в рамках параметров, установленных при их аттестации (испытаниях).
- Следует предусмотреть меры предосторожности для предотвращения потенциального заражения вирусами, начиная со стадий до и после удаления (инактивации) вирусов. Открытая обработка должна проводиться в зонах, отделенных от других технологических операций и имеющих отдельную систему вентиляции.
- Как правило, для различных стадий очистки не используют одно и то же оборудование. Если оборудование используют для разных стадий очистки, то перед повторным использованием оно должно быть очищено и дезинфицировано соответствующим образом. Следует предпринимать меры, предотвращающие перенос вирусов с предыдущих стадий (например, через оборудование или окружающую среду).
19. АФС, предназначенные для проведения клинических исследований
19.1 Общие положения
-
- При производстве новых АФС, используемых для проведения клинических исследований, при их разработке могут применяться не все виды контроля, рассмотренные в предыдущих разделах данного стандарта. В настоящем разделе приведены специальные требования к этим АФС.
- Контроль производства АФС, предназначенных для клинических исследований, должен соответствовать той стадии разработки лекарственного средства, в состав которого она входит. Методы производства и контроля качества таких АФС должны быть достаточно гибкими для того, чтобы предусмотреть изменения, вносимые в них по мере получения новых знаний о технологическом процессе и продвижении лекарственного средства с доклинических стадий к клиническим стадиям исследований. Если разработка лекарственного препарата достигает стадии, на которой эта АФС про-
- Руководство ICH Q5A «Качество биотехнологических продуктов. Оценка безопасности вирусного заражения биотехнологических продуктов, произведенных из клеток человеческого и животного происхождения» (Quality of Biotechnological Products: Viral Safety Evaluation of Biotechnology Products Derived from Cell Lines of Human or Animal Origin).
изводится для применения в лекарственном препарате, предназначенном для проведения клинических исследований, производители должны гарантировать, что производственные мощности, соответствующие методы производства и контроля обеспечивают необходимое качество этой АФС.
19.2 Качество
-
- К производству АФС, предназначенных для использования при проведении клинических исследований, должны применяться требования настоящего стандарта с надлежащей процедурой выдачи разрешения на реализацию каждой серии.
- Для выдачи разрешения на реализацию или отклонение каждой серии АФС, предназначенной для использования в клинических исследованиях, должен(ы) быть образован(ы) отдел(ы) контроля качества, независимый(е) от производства.
- Отдельные работы по проведению контроля, обычно выполняемые отделом контроля качества, могут проводиться в других подразделениях.
- Мероприятия по контролю качества должны включать в себя проведение контроля исходных и упаковочных материалов, а также промежуточных продуктов и АФС.
- Следует проводить анализ любых затруднений, связанных с производством и качеством продукции.
- Маркировка АФС, используемых в клинических исследованиях, должна контролироваться установленным образом и содержать информацию о том, что это вещество предназначено для исследовательских целей.
19.3 Оборудование
-
- При проведении всех стадий клинических разработок (в т. ч. при использовании опытных участков или лабораторий) для производства АФС, предназначенных для использования в клинических исследованиях, должен быть установлен порядок проведения калибровки и очистки оборудования и методики оценки его соответствия своему назначению.
- Порядок эксплуатации оборудования должен гарантировать, что при работе с материалами риск загрязнения и перекрестного загрязнения будет минимальными.
19.4 Контроль исходных материалов
-
- Исходные материалы, применяемые для производства АФС, предназначенные для использования в клинических исследованиях, должны быть испытаны или должны иметь аналитический паспорт поставщика и проверяться на подлинность. Наличие аналитического паспорта поставщика достаточно для подтверждения качества материала, представляющего опасность.
- В некоторых случаях подтверждение пригодности исходного материала для его использования может быть обосновано не только результатами аналитического контроля, но и упрощенными методами (например, проверкой пригодности).
19.5 Производство
-
- Процесс производства АФС, предназначенных для использования в клинических исследованиях, должен быть зарегистрирован в лабораторных журналах, протоколах серии или путем использования других средств. Эти документы должны включать в себя информацию об использовании технологических материалов, оборудовании, технологическом процессе и научных наблюдениях.
- Ожидаемые выходы продукции могут различаться более значительно и быть менее определенными, чем ожидаемые выходы продукции процессов, выполняемых в промышленном масштабе. Нет необходимости проводить изучение различия выходов продукции.
19.6 Аттестация (испытания)
-
- Как правило, не требуется проводить аттестацию (испытания) процесса производства АФС, предназначенных для использования в клинических исследованиях, если производится одна серия АФС или из-за изменений, вносимых в технологический процесс при разработке АФС, повторное изготовление серии является затруднительным или неточным. В этот период разработки качество АФС обеспечивается сочетанием контроля, калибровки (поверки) и аттестации оборудования.
- Аттестацию процесса в соответствии с разделом 12 настоящего стандарта следует проводить для серий АФС, предназначенных для реализации, даже если такие серии производятся на опытном или мелкомасштабном производстве.
19.7 Изменения
19.70 Изменения следует вносить по мере приобретения новых знаний и роста масштаба производства. Каждое изменение технологии производства, спецификаций или методик испытаний должно быть зарегистрировано установленным образом.
19.8 Лабораторный контроль
-
- Несмотря на то, что аналитические методы, используемые для оценки качества серии АФС, предназначенных для клинических исследований, не могут быть аттестованы, они должны быть научно обоснованы.
- Должна быть организована система хранения архивных образцов всех серий АФС, обеспечивающая хранение каждого из архивных образцов в достаточном количестве и надлежащих условиях в течение установленного периода времени после получения разрешения на реализацию, завершения клинического исследования или отзыва заявки на новое лекарственное средство.
- Срок годности и дата повторного контроля, указанные в 11.6 настоящего руководства, применимы к существующим АФС, предназначенным для использования в клинических исследованиях. Для новых АФС, находящихся на ранних стадиях клинических исследований, положения 11.6, как правило, применимы.
19.9 Документация
-
- Система документации должна гарантировать, что информация, полученная в ходе разработки и производства АФС, предназначенных для использования в клинических исследованиях, будет оформлена документально должным образом и доступна для использования.
- Разработка и применение аналитических методов, используемых для подтверждения выпуска серии АФС, предназначенной для клинических исследований, должны быть оформлены документально.
- Должен быть разработан и внедрен порядок хранения протоколов производства и контроля качества АФС и соответствующей документации. Этот порядок должен обеспечивать хранение протоколов и документов в течение установленного периода времени после получения разрешения на реализацию, завершения клинического исследования или отзыва заявки на новое лекарственное средство.
20. Термины и определения
активная фармацевтическая субстанция; АФС (active pharmaceutical ingredient; API): Любое вещество или смесь веществ, предназначенные для производства лекарственных средств, которые в процессе производства лекарственного средства становятся активным ингредиентом этого лекарственного средства. Такие вещества предназначены для проявления фармакологической активности или другого прямого эффекта при диагностике, лечении, облегчении симптомов или профилактике болезни или для воздействия на структуру или функцию организма.
аттестация (qualification): Документальное подтверждение того, что методика, процесс, оборудование, материал или система соответствуют заданным требованиям, и их использование дает ожидаемые результаты.
биологическая нагрузка (bioburden): Уровень и тип (например, патогенных или непатогенных) микроорганизмов, которые могут присутствовать в сырье, исходных материалах АФС, промежуточных продуктах или АФС. Биологическая нагрузка не рассматривается как загрязнение, если ее уровни не превышают установленные предельные значения или не происходит обнаружения определенных патогенных микроорганизмов.
внутрипроизводственный контроль (технологический контроль) (in process control (or process control): Контроль, проводимый в ходе технологического процесса с целью проверки соответствия промежуточного продукта или АФС установленным требованиям, по результатам которого могут корректироваться параметры технологического процесса.
вспомогательные средства (process aids): Материалы (за исключением растворителей), используемые в качестве добавок при производстве промежуточного продукта или АФС, которые сами не участвуют в химической или биологической реакции (например, порошок для фильтрования, активированный уголь и т.д.).
выход ожидаемый (yield, expected): Количество или процент теоретического выхода материала, ожидаемый на любой соответствующей стадии производства, определяемый на основании данных, предварительно полученных при производстве этого материала в лабораторных, опытных или промышленных условиях.
выход теоретический (yield, theoretical): Количество, которое было бы получено на любой соответствующей стадии производства, рассчитанное на основе количества используемого материала в предполагаемых условиях отсутствия любых потерь или ошибок при фактическом производстве.
дата повторного контроля (retest date): Дата прохождения материалом повторной проверки для подтверждения его пригодности для дальнейшего использования.
загрязнение (contamination): Нежелательное внесение примесей химического или микробиологического происхождения или постороннего материала в исходный материал, промежуточный продукт или АФС в ходе производства, отбора проб, упаковки или переупаковки, хранения или транспортирования.
инструкция, методика, процедура (procedure): Документ, содержащий указания по выполнению отдельных видов операций (например, по очистке, переодеванию, контролю окружающей среды, отбору проб, проведению испытаний, эксплуатации оборудования), который прямо или косвенно связан с производством промежуточного продукта или АФС.
испытания (validation): Синоним термина «аттестация».
исходный материал АФС (API starting material): Сырье, промежуточный продукт или АФС, которые используются при производстве АФС и внедряются в структуру АФС в качестве существенного элемента. Исходный материал АФС может быть продуктом (материалом), получаемым от одного или более поставщиков по контракту (коммерческому соглашению), или материалом, производимым на самом предприятии. Как правило, для производства АФС используются исходные материалы с конкретными химическими свойствами и структурой.
калибровка (поверка) (calibration): См. общие термины и определения.
карантин (quarantine): Статус материалов, изолированных физически или иным образом на время до вынесения решения об их выпуске или отклонении.
контроль качества (quality control; QC): Проведение проверок или испытаний на соответствие требованиям спецификаций.
критерии приемлемости (acceptance criteria): Числовые предельные значения, диапазоны или другие критерии, применяемые для приемки результатов испытаний.
критический (critical): Стадии технологического процесса, требования к испытаниям, существенным параметрам или условиям, которые следует задать и контролировать для обеспечения соответствия АФС требованиям спецификации.
лекарственная субстанция (drug substance): Синоним термина «активная фармацевтическая субстанция»
лекарственный (медицинский) препарат (drug (medicinal) product): Дозированная форма лекарственного средства в первичной окончательной упаковке, предназначенной для продажи.
материал (material): Сырье (исходные материалы, реактивы, растворители), технологические добавки, промежуточные продукты, АФС, упаковочные и печатные материалы.
маточный раствор (mother liquor): Остаточная жидкость после процессов кристаллизации и выделения.
П р и м е ч а н и е – Маточный раствор может содержать непрореагировавшие материалы, промежуточные продукты, АФС и/или примеси в существенном количестве, а также использоваться для дальнейшей обработки.
методика аттестации (испытаний) (validation protocol): Документально оформленный план, устанавливающий порядок аттестации (испытаний) и определяющий критерии приемлемости.
П р и м е ч а н и е – Например, методика аттестации (испытаний) технологического процесса, конкретно определяющая технологическое оборудование, критические технологические параметры (рабочие диапазоны), характеристики продукции, отбор проб, данные испытаний, которые должны быть собраны, количество циклов аттестации (испытаний) и приемлемые результаты испытаний.
номер серии (партии) (batch number or lot number): Определенное сочетание цифр, букв и/или символов, обозначающее серию продукции. По номеру серии может быть прослежен весь процесс производства серии АФС и ее реализации.
обеспечение качества (quality assurance; QA): Общая совокупность организационных мероприятий, проводимых с целью обеспечения требуемого качества всех АФС и поддерживания систем качества на должном уровне.
Служба (отдел) качества (quality unit(s): Организационное подразделение, независимое от производства, которое несет ответственность за обеспечение и контроль качества, представляющее собой отделы контроля и обеспечения качества либо одно лицо или группу, в зависимости от размера и структуры организации.
отклонение (deviation): Отступление от утвержденной инструкции или установленного стандарта.
перекрестное загрязнение (cross-contamination): Загрязнение материала или продукта другим материалом или продуктом.
переработка (reworking): Обработка промежуточного продукта или АФС, не удовлетворяющих требованиям стандартов или спецификаций, с помощью одной или нескольких технологических стадий, отличающихся от установленного технологического процесса, с целью получения промежуточного продукта или АФС приемлемого качества (например, перекристаллизация с другим растворителем).
повторная обработка (reprocessing): Повторное введение промежуточного продукта или АФС (в т. ч. не удовлетворяющих требованиям спецификаций) в процесс и повторение стадии кристаллизации или другой соответствующей стадии химической или физической обработки (например, дис-
тилляции, фильтрации, хроматографии, измельчения), которая является частью установленного технологического процесса. Продолжение технологической стадии после того как внутрипроизводственный контроль показал, что она еще не завершена, считается частью текущего процесса производства и не рассматривается как повторная обработка.
подпись (согласование) (signature (signed): См. термин «согласование».
подрядчик (contract manufacturer): Производитель, выполняющий некоторые производственные операции от имени первоначального производителя.
примесь (impurity): Компонент, присутствующий в промежуточном продукте или АФС, который является нежелательным для этого продукта.
производство (manufacture): Операции и виды контроля, связанные с закупкой исходных материалов, их приемкой, обработкой, упаковкой, выпуском в реализацию, хранением и отгрузкой АФС.
промежуточный продукт (intermediate): Материал, получаемый на стадиях производства АФС, который претерпевает дальнейшие молекулярные изменения или очистку до того, как он может считаться АФС. Промежуточные продукты могут не выделяться в качестве чистых продуктов.
П р и м е ч а н и е – Настоящий стандарт распространяется только на промежуточные продукты, производство которых начинается со стадии, являющейся, по документам производителя, началом производства АФС.
растворитель (solvent): Неорганическая или органическая жидкость, используемая в качестве носителя для приготовления растворов или суспензий при производстве промежуточного продукта или АФС.
серия (партия) (batch or lot): Определенное количество материалов, произведенных в одном или нескольких технологических процессах так, что их можно считать однородными в рамках установленных пределов. При непрерывном производстве понятие серии может относиться к определенной части продукции, характеризуемой однородностью. Размер серии может быть установлен по фиксированному количеству продукции либо по количеству продукции, производимой за установленный период времени.
система с компьютерным управлением и контролем (computerized system): Система, включающая в себя ввод данных, их электронную обработку и вывод информации, используемой для регистрации или автоматического контроля.
согласование (подпись) (signed signature): Подпись лица, выполняющего конкретное действие или обзор, в виде сокращенной или полной фамилии и инициалов, сделанной от руки, личной печати или авторизованной и защищенной электронной подписи.
состав примесей (impurity profile): Описание идентифицированных и неидентифицированных примесей, присутствующих в АФС.
спецификация (specification): Перечень испытаний, ссылок на аналитические методики и соответствующие критерии приемлемости, устанавливающие числовые границы, диапазоны или другие критерии для указанных испытаний. Спецификация устанавливает критерии, которым должен соответствовать материал для того, чтобы его можно было применять по назначению. Материал соответствует требованиям спецификации, если при испытаниях по принятым аналитическим методам он удовлетворяет установленным критериям приемлемости.
срок годности (expiry date or expiration date): Дата, указываемая на упаковке/этикетке АФС, обозначающая период времени, в течение которого гарантируется сохранение свойств АФС в рамках установленных спецификаций при хранении в определенных условиях и после которого данная АФС не должна использоваться.
сырье (raw material): Исходные материалы, реактивы и растворители, предназначенные для изготовления промежуточного продукта или АФС.
технологический контроль (process control): См. термин «внутрипроизводственный контроль».
технологический процесс (production): Операции, включающие в себя приемку исходных материалов, их обработку, упаковку и получение готовой АФС.
упаковочный материал (packaging material): Любой материал, предназначенный для защиты промежуточного продукта или АФС при хранении и транспортировании.
эталонный стандарт, вторичный (reference standard, secondary): Вещество установленного качества и чистоты по результатам сравнения с первичным эталонным стандартом, используемое в качестве стандарта сравнения для текущих лабораторных анализов.
эталонный стандарт, первичный (reference standard, primary): Вещество, признанное подлинным материалом высокой степени чистоты по результатам обширных аналитических испытаний. Этот стандарт может быть: 1) получен из официально признанного источника; 2) приготовлен путем независимого синтеза; 3) получен из имеющегося промышленного материала высокой степени чистоты или 4) приготовлен путем дальнейшей очистки существующего промышленного материала.
1. Организация работы по обеспечению качества
Общие положения
Производитель лекарственных средств (держатель регистрационного досье) должен произво- дить их так, чтобы лекарственные средства гарантированно соответствовали своему назначению, регистрационному досье или протоколу клинических испытаний и соответственно не создавали риск для потребителей из-за нарушения условий безопасности, качества или эффективности. Достижение этой цели возлагает ответственность на руководителей предприятия и требует участия и инициативы работников многих подразделений на всех уровнях предприятия, а также поставщиков и дистрибью- торов. Для надежного достижения этой цели должна действовать детально разработанная и пра- вильно применяемая фармацевтическая система качества1, включающая правила производства и контроля качества и анализ рисков, которые должны быть полностью оформлены документально и эффективность которых следует проверять. Все составляющие фармацевтической системы качества должны быть обеспечены компетентным персоналом, требуемыми помещениями и оборудованием. Дополнительная устанавливаемая законодательством ответственность возлагается на держателей регистрационного досье и уполномоченных лиц.
Основные принципы обеспечения качества, Правил GMP, контроля качества и анализ рисков взаимосвязаны. Они приведены в настоящем стандарте и имеют первостепенное значение в органи- зации производства лекарственных средств.
Фармацевтическая система качества1
-
- Обеспечение качества (управление качеством) является комплексной задачей, охватываю- щей все факторы, которые по отдельности или совместно влияют на качество продукции. Оно явля- ется итогом всех мер, предпринимаемых с целью обеспечения соответствия лекарственных средств своему назначению. Система качества включает в себя правила производства и контроля качества.
1.2. Область применения правил GMP включает в себя такие стадии жизненного цикла как про- изводство лекарственных средств для исследований, передачу технологии, коммерческое производ- ство до прекращения выпуская продукции. Однако область применения фармацевтической системы качества может быть расширена на разработку лекарственного средства согласно руководству ICH Q10, которое носит рекомендательный характер, но может способствовать развитию и постоянному улучшению и усиливать связь между стадиями разработки и производства. Руководство ICH Q10 при- ведено в части III стандарта и может служить дополнением к данной главе.
-
- При разработке новой фармацевтической системы качества или совершенствовании суще- ствующей следует учитывать масштаб и сложность деятельности предприятия. Разработка системы должна включать необходимый анализ рисков с использованием соответствующих методов. В то время как некоторые элементы системы могут относиться ко всему предприятию, а другие – к опре- деленному производству, эффективность системы обычно оценивается на уровне производства.
- Система обеспечения качества (система качества) при производстве лекарственных средств должна гарантировать следующее:
- Производство продукции основано на разработке, внедрении, поддержании и непрерывном совершенствовании системы, которая обеспечивает постоянную реализацию продукции в соответст- вии с требованиями к качеству;
- Знания о продукте и процессе поддерживаются на всех этапах жизненного цикла;
- Лекарственные средства разработаны с учетом требований настоящего стандарта; (iv)Производственные и контрольные операции четко специфицированы в соответствии с тре-
бованиями настоящего стандарта;
- Ответственность работников четка определена;
- Предусмотрены меры, обеспечивающие производство, поставку и использование нужных исходных и упаковочных материалов, выбор и контроль поставщиков и проверку того, что каждая по-
1 Раздел 6 Директивы 2003/94/ЕС и 91/412/ЕЕС требуют от производителей разработать и применять эффектив- ную фармацевтическую систему обеспечения качества. Термин «Фармацевтическая система качества» используется в этой главе с целью преемственности терминологии ICH Q10. Для целей данной главы оба термина являются взаимо- заменяемыми.
ставка выполнена согласно утвержденной схеме снабжения;
- Существует схема организации и контроля работ по контракту;
- Введен и выполняется контроль за счет разработки и использования эффективных систем контроля за процессом и качеством продукции;
- При выпуске серий продукции учитываются результаты контроля процессов и продукции, проводятся расследования отклонений и выполняются предупредительные меры во избежание от- клонений в будущем;
- Предусмотрен контроль промежуточной продукции и технологического процесса (внутрипро- изводственный контроль), аттестация (испытания) процессов и оборудования проводятся в необхо- димом объеме;
- Проводится постоянное совершенствование путем улучшения характеристик качества, соот- ветствующее знаний о процессе и продукции;
- Предусмотрена перспективная оценка планируемых изменений и их утверждение с учетом уведомления надзорных органов и согласования (утверждения) при необходимости;
- Проведение оценки после введения любых изменений для подтверждения качества и от- сутствия нежелательного влияния на качество продукции;
- При расследовании следует установить причину отклонений, предполагаемых дефектов продукции и в других случаях. Это может быть сделано с помощью анализа рисков. В случае, если истинную причину найти не удается, следует принять во внимание наиболее вероятную причину и работать с ней. Если допускается или установлена ошибка человека в качестве причины, то это должно быть обосновано, причем нельзя пропустить технологические, методические или системные ошибки, если они имеют место. Результатом расследования должны быть разработка и реализация предупредительных и корректирующих действий (САРА). Следует проверять и оценивать эффектив- ность таких действий, совместно с принципами анализа рисков;
- Не допускается реализация лекарственных средств до выдачи уполномоченным лицом разрешения на выпуск, которое должно подтвердить, что каждая серия продукции произведена и проверена в соответствии с требованиями регистрационного досье и другими требованиями к произ- водству, контролю и реализации лекарственных средств;
- Существующая система мер обеспечивает качество лекарственных средств при их хране- нии, отгрузке и последующем обращении в течение всего срока годности;
- Порядок проведения самоинспекции и/или аудита качества позволяет регулярно оцени- вать эффективность фармацевтической системы качества.
-
- Руководство предприятия несет полную ответственность за эффективность фармацевтиче- ской системы качества, обеспечение ее необходимыми ресурсами, распределением обязанностей, ответственности и прав и охват ею всего предприятия. Важную роль играет руководящая роль руко- водства предприятия и его активное участие в фармацевтической системе качества. Это должно обеспечивать поддержку и участие персонала на всех уровнях и участках предприятия в фармацев- тической системе качества.
- Следует периодически проводит анализ фармацевтической системы качества с участием руководства предприятия для определения возможностей постоянного улучшения продукции, про- цессов и самой системы.
- Следует создать и документально оформить фармацевтическую систему качества. Следует разработать руководство по качеству или эквивалентный ему документ, содержащий описание сис- темы обеспечения качества, включая ответственность персонала.
Правила производства и контроля качества лекарственных средств
-
- Правила производства и контроля качества являются частью системы обеспечения качеств, которые обеспечивают непрерывное производство и контроль качества продукции в соответствии со стандартами качества исходя из назначения продукции и согласно регистрационному досье, протоко- лу клинических испытаний или спецификации на продукцию. Правила распространяются как на про- изводство, так и на контроль. Основные требования правил заключаются в следующем:
- Все производственные процессы должны быть четко регламентированы и должны периоди- чески пересматриваться с учетом накопленного опыта. Следует показывать, что они могут постоянно производить лекарственные средства требуемого качества в соответствии со спецификациями;
- Следует проводить аттестацию (испытания) критических стадий процессов производства, в том числе при внесении существенных изменений в технологический процесс;
- Существуют все необходимые помещения и оборудования с необходимым обеспечением в соответствии с GMP, включая:
- обученный и аттестованный персонал;
- необходимые помещения и площади;
- соответствующее оборудование и системы обслуживания;
- материалы, средства упаковки и маркировки, удовлетворяющих установленным требованиям;
- утвержденные инструкций и методики в соответствии с фармацевтической системой качества;
- требуемые условия хранения и транспортирования;
- Инструкции и методики должны быть конкретными, изложены ясно и однозначно в письмен- ной форме для предмета, к которому они относятся;
- Персонал должен быть обучен правильному выполнению инструкций;
- В процессе производства следует составлять протоколы (заполняемые рукописным спосо- бом и/или с применением технических средств), документально подтверждающие фактическое вы- полнение предусмотренных инструкциями технологических стадий и получение продукции требуемо- го качества и в заданном количестве;
- Все существенные отклонения следует протоколировать в полном объеме и расследовать с целью установления их причины и принятия необходимых предупреждающих и корректирующих действий;
- Протоколы на серию продукции, в т. ч. на документацию по реализации продукции, должны давать возможность прослеживать изготовление каждой серии и храниться в полном объеме в дос- тупной форме;
- Порядок реализации (оптовой продажи) продукции должен сводить к минимуму любой риск для ее качества в соответствии с правилами обращения в системе оптовой торговли;
- Следует организовать систему отзыва любой серии продукции из продажи или поставки;
- Рекламации на качество продукции следует тщательно рассматривать, а причины ухудше- ния качества расследовать с принятием соответствующих мер по их предотвращению.
Контроль качества
-
- Контроль качества является частью настоящего стандарта (правил GMP) и включает в себя отбор проб, проведение испытаний (анализов) и оформление соответствующей документации. Инст- рукции по организации, документированию и выдаче разрешения на выпуск продукции должны вклю- чать в себя проведение всех необходимых испытаний и запрещать использование исходного сырья и материалов и реализацию готовой продукции до подтверждения соответствия качества установлен- ным требованиям. Основные требования к контролю качества:
- Наличие необходимых помещений и оборудования, обученного персонала, утвержденных методик по отбору проб, проверке и проведению испытаний исходных и упаковочных материалов, промежуточной, нерасфасованной и готовой продукции, контролю окружающей среды, при необходи- мости, в соответствии с требованиями настоящего стандарта (правил GMP);
- Проведение отбора проб исходных и упаковочных материалов, промежуточной, нерасфасо- ванной и готовой продукции аттестованным персоналом в соответствии с утвержденными методиками;
- Проведение испытаний аттестованными методами;
- Составление протоколов (заполняемых рукописным способом и/или с применением техни- ческих средств), подтверждающих фактическое проведение всех необходимых отборов проб, прове- рок и испытаний, а также регистрацию любых отклонений и расследований в полном объеме;
- Подтверждение того, что готовая продукция содержит активные фармацевтические субстан- ции (ингредиенты), по качественному и количественному составу соответствующие регистрационному досье или протоколу клинических испытаний, имеет требуемую чистоту, правильно упакована и мар- кирована;
- Оформление протоколов проверки исходного сырья и материалов, промежуточной, нерас- фасованной и готовой продукции, их анализ и сравнение со спецификациями. Оценка продукции включает в себя рассмотрение всей необходимой производственной документации и анализ отклоне- ний от установленных требований;
- Получение разрешения на продажу или поставку любой серии продукции только после подтверждения уполномоченным лицом ее соответствия требованиям, установленным при государ- ственной регистрации в соответствии с приложением 16;
- Сохранение достаточного количества образцов исходных материалов и продукции в соот- ветствии с приложением 19 для возможной проверки в случае необходимости. Образцы продукции следует хранить в их окончательной упаковке
Анализ качества продукции
-
- Следует регулярно проводить анализ качества продукции и оформлять его документально в виде аналитического обзора для всех лицензированных лекарственных средств, включая предна- значенные только для экспорта, с целью проверки неизменности действующих процессов, соответст- вия спецификациям исходных материалов и готовой продукции для обнаружения любых изменений и тенденций и подтверждения совершенствования продукции и процессов. Как правило, такой анализ следует, как правило, проводить один раз в год, принимая во внимание результаты проведенных ра- нее анализов.
Проведение анализа должно включать в себя, как минимум, следующее:
- Анализ исходных и упаковочных материалов, используемых для производства продукции, обращая особое внимание на материалы, полученные от новых поставщиков прослеживаемость цепи снабжения активных субстанций;
- Анализ критических параметров при внутрипроизводственном контроле и контроле готовой продукции;
- Анализ всех серий, для которых отмечены отклонения от спецификаций, и результатов рас- следования этих отклонений;
- Анализ всех существенных отклонений или несоответствий, результатов их расследования и оценка эффективности предпринимаемых корректирующих и предупреждающих мер;
- Анализ всех изменений, вносимых в процессы или аналитические методы;
- Анализ изменений в регистрационной документации (представленных, принятых или откло- ненных), включая изменения, относящиеся к продукции, предназначенной только для экспорта;
- Анализ результатов контроля стабильности и любых отрицательных тенденций;
- Анализ возвратов продукции, связанных с качеством, рекламаций и отзывов продукции и результатов проведенных по ним расследований;
- Анализ эффективности любых ранее внесенных изменений в процессы или оборудование;
- Анализ замечаний на вновь зарегистрированные лекарственные средства или после внесе- ния изменений в документы, относящиеся к государственной регистрации; анализ обязательств после реализации;
- Рассмотрение результатов аттестации (испытаний) оборудования, процессов и технологиче- ских сред, например, систем вентиляции и кондиционирования воздуха, подготовки воды, сжатого воздуха и др.;
- Рассмотрение работы по контрактам (раздел 7) с целью подтверждения их соответствия реальной практике.
1.11. Производитель и владелец регистрационного свидетельства (лицензии на производство), если они являются разными лицами, должны оценить результаты данного анализа, установить несо- ответствия и дать предложения по принятию корректирующих и предупреждающих действий и необ- ходимости проведения повторной аттестации (испытаний) в соответствии с фармацевтической сис- темой качества. Для выполнения этих требований и контроля результатов должны быть разработаны инструкции и должна быть проверена эффективность принятых мер в ходе самоинспекции. Проведе- ние анализа качества может выполняться отдельно для разных видов продукции, например, твердых форм, жидких форм, стерильной продукции и т. д., при наличии обоснования.
Если владелец регистрационного досье не является производителем продукции, то в специ- альном соглашении между этими сторонами должна быть определена ответственность каждой сто- роны в отношении проведения анализа качества.
Анализ рисков
-
- Анализ рисков является систематизированным процессом оценки, принятия решений и мер, связанных с рисками, влияющими на качество лекарственного средства. Он может проводиться как в перспективном, так и в ретроспективном (на основе предшествующих данных) плане.
- Анализ рисков основан на следующих принципах:
- оценке риска для качества, основываясь на научном подходе, опыте работы и, в конечном счете, исходя из задачи защиты потребителя;
- масштаб работы, степень формализации и документального оформления процесса анализа риска соответствуют уровню риска.
Примеры анализа рисков и области применения могут быть найдены в разных источниках, в т. ч. в руководстве ICH Q9, которые содержится в части III стандарта.
2.Персонал
Основные принципы
Производство лекарственных средств в соответствии с установленными требованиями зависят от персонала. Для выполнения всех задач, входящих в область ответственности предприятия, оно должно быть укомплектовано персоналом необходимой квалификации. Должностные обязанности каждого сотрудника должны быть оформлены документально и усвоены им. Все сотрудники должны знать требования настоящего стандарта (правил GMP), относящиеся к сфере их деятельности, и пройти начальное и повторное обучение в соответствии с их обязанностями, в т. ч. по правилам лич- ной гигиены.
Общие положения
-
- Предприятие должно быть укомплектовано достаточным количеством персонала, имеющим необходимую квалификацию и практический опыт работы. Руководство предприятия должно опреде- лить необходимые ресурсы (людские, финансовые, материальные, иметь помещения и оборудова- ние) для внедрения и непрерывного повышения ее эффективности. Должностные обязанности от- дельного сотрудника не должны быть слишком большими, что может привести к риску для качества продукции.
- Предприятие должно иметь четкую организационную структуру, отражающую взаимосвязи руководителей производства, подразделения контроля качества и, если предусмотрено, руководите- ля подразделения или отдела обеспечения качества согласно п. 2.5 и на которой четко показано ме- сто уполномоченного лица (лиц) в системе управления.
- Служебные обязанности руководителей и других ответственных работников должны быть изложены в должностных инструкциях. Эти лица должны иметь достаточные полномочия для выпол- нения своих функций. Их полномочия могут быть переданы официально назначенным заместителям, имеющим достаточную квалификацию. Следует исключить неоправданное дублирование ответствен- ности сотрудников, связанной с выполнением требований настоящего стандарта (GMP), и не допус- кать случаев, когда какие-либо функции ни за кем не закреплены.
- Руководство предприятия несет ответственность за за обеспечение функционирования эф- фективной системы качества для достижения целей качества и что роли, ответственность и полномо- чия определены, взаимосвязаны и внедрены на всем предприятии. Руководство предприятия должно ввести политику качества, которая определяет общее направление работы в области качества, ее постоянное соответствие требованиям и эффективность системы качества и соответствие правилам GMP за счет участия в рассмотрении работы предприятия.
Руководящие работники
-
- Руководство предприятия должно назначить руководителей, включая руководителей произ- водства, подразделения контроля качества и, если хотя бы один из этих руководителей не выполняет обязанности согласно статье 51 Директивы 2001/83/ЕС, должны быть назначены уполномоченные лица в требуемом количестве, но не менее одного, для выполнения этих функций.
Основные руководители должны быть заняты на предприятии, как правило, полный рабочий день. Руководители производства и подразделения контроля качества должны быть независимыми друг от друга. На больших предприятиях часть функций, перечисленных в 2.7–2.9, допускается, при необходимости, передавать другим сотрудникам. Дополнительно, в зависимости от размеров и струк- туры компании, могут назначаться отдельно руководители подразделений контроля и обеспечения качества. В этом случае некоторые обязанности по п.п. 2.7–2.9 разделяются между руководителями подразделения контроля качества и производства и руководство предприятия должно проследить, чтобы их ответственность и полномочия были четко определены.
-
- Обязанности уполномоченных лиц заключаются в следующем:
- В отношении лекарственных средств, выпущенных в Российской Федерации, уполномочен- ное лицо должно гарантировать, что каждая серия продукции была изготовлена и проверена в соот- ветствии с установленными требованиями регистрационного досье.
- В отношении лекарственных средств, выпущенных за пределами Российской Федерации, уполномоченное лицо должно гарантировать, что каждая импортируемая серия продукции прошла полный количественный анализ, количественный анализ всех активных субстанций и все другие тес- ты, необходимые для обеспечения качества лекарственных средств в соответствии с требованиями регистрационного досье и в порядке, установленном в Российской Федерации. Уполномоченное лицо должно указать в реестре или эквивалентном ему документе, что эти проверки выполнены и удосто- вериться до выпуска каждой серии, что она соответствует в порядке, установленном в Российской Федерации.
Лица, выполняющие эти обязанности, должны удовлетворять требованиям к квалификации в порядке, установленном в Российской Федерации, и должны находиться постоянно в распоряжении держателя регистрационного досье для выполнения своих обязанностей.
Ответственность уполномоченного лица может быть передана только другому уполномоченно- му лицу.
Руководство по работе уполномоченных лиц приведено в приложении 16.
-
- На руководителя производства возлагаются, как правило, следующие обязанности:
- Организация производства и хранения продукции в соответствии с документацией с целью обеспечения требуемого качества;
- Утверждение инструкций, связанных с производственным процессом, и обеспечение их точ- ного выполнения;
- Обеспечение рассмотрения и подписания всех производственных протоколов лицами,
имеющими необходимые полномочия;
- Обеспечение аттестации (испытаний) и обслуживания помещений и оборудования в своем подразделении;
- Контроль проведения работ по аттестации (испытаниям) процессов;
- Контроль за организацией первичного и последующего обучения производственного персо- нала в соответствии с требованиями.
-
- Основные обязанности руководителя подразделения контроля качества состоят в следующем:
- Утверждение или отклонение исходных и упаковочных материалов, промежуточной, нерас- фасованной и готовой продукции.
- Обеспечение проведения всех предусмотренных проверок и испытаний и оценки связанных с ними протоколов;
- Утверждение спецификаций, инструкций по отбору проб, методик испытаний и других доку- ментов по контролю качества;
- Допуск к работе специалистов аналитиков, работающих по контракту, и контроль их дея- тельности;
- Обеспечение работы своего подразделения, обслуживания его помещений и оборудования;
- Контроль проведения аттестации (испытаний);
- Организация первичного и последующего обучения персонала своего подразделения и ве- дение соответствующих протоколов.
Другие обязанности сотрудников подразделения контроля качества приведены в разделе 6.
-
- Руководители производства и подразделения контроля качества и, где требуется подразде- ления обеспечения качества, имеют ряд раздельных и совместных обязанностей, относящихся к ка- честву, включая, в особенности, разработку, эффективное применение, контроль и поддержание сис- темы качества. Эти обязанности с учетом действующих норм и правил могут включать в себя сле- дующее:
- утверждение письменных инструкций, методик и других документов, в т. ч. внесение в них изменений;
- контроль производственной среды;
- контроль соблюдения правил производственной гигиены;
- аттестация (испытания) процессов;
- обучение персонала;
- утверждение и контроль поставщиков материалов;
- утверждение и контроль производителей, работающих по контракту, и других партнеров, связанных с правилами GMP;
- определение условий хранения материалов, продукции и контроль их соблюдения;
- хранение протоколов;
- контроль соответствия требованиям настоящего стандарта;
- проведение инспекций, расследований и отборов проб с целью выявления факторов, спо- собных повлиять на качество продукции
- Участие в анализе процессов, качества продукции и системы обеспечения качества и под- держка постоянного совершенствования;
- Обеспечение своевременного и эффективного взаимодействия и совершенствования с це- лью улучшения показателей качества до необходимого уровня управления.
Обучение
-
- Предприятие-производитель должно обеспечить обучение всех сотрудников, занятых произ- водством или контролем качества (в т. ч. технический, обслуживающий персонал и персонал, выполняю- щий уборку), а также других сотрудников, деятельность которых может влиять на качество продукции.
- Помимо базового обучения по теории и практике системы обеспечения качества и требо- ваниям настоящего стандарта (GMP) вновь принятые сотрудники должны пройти обучение, соответ- ствующее их должностным обязанностям. Следует организовать периодическое обучение персонала и оценивать эффективность этого обучения на практике. Обучение следует проводить по програм- мам, утвержденным руководителями производства или подразделения контроля качества. На пред- приятии должна храниться документация о проведении обучения.
- Сотрудники, работающие в зонах, в которых загрязнение представляет опасность, напри- мер, в чистых зонах или в зонах работы с сильнодействующими, токсичными, инфицирующими или сенсибилизирующими веществами, должны пройти специальное обучение.
- Посетители и/или необученные сотрудники не должны, как правило, допускаться в зоны, связанные с производством и контролем качества. При необходимости, они должны предварительно пройти инструктаж по правилам личной гигиены, порядку переодевания и ношению специальной оде- жды. За этими лицами должен быть организован тщательный контроль.
- При обучении следует подробно разъяснять принципы системы обеспечения качества и
меры по ее улучшению продукции для их полного усвоения и дальнейшего применения.
Гигиена персонала
-
- На предприятии должны быть разработаны и внедрены инструкции по личной гигиене пер- сонала с учетом особенностей конкретного производства. Инструкции должны устанавливать требо- вания к состоянию здоровья сотрудников, соблюдению ими личной гигиены и правилам ношения одежды. Эти инструкции должны понимать и строго соблюдать все сотрудники, связанные с нахож- дением в производственных помещениях и помещениях контроля качества. Руководство предприятия несет ответственность за выполнение персоналом правил личной гигиены и организацию необходи- мого обучения.
- Все лица, принимаемые на работу, должны пройти медицинский осмотр. На предприятии должны быть разработаны и внедрены инструкции с перечнем показателей состояния здоровья, ко- торые могут оказать влияние на качество продукции и о которых должны знать работодателя. В слу- чаях, связанных с производственной необходимостью или состоянием здоровья, сотрудники должны проходить повторный медицинский осмотр.
- Лица с инфекционными заболеваниями и повреждениями на открытых участках тела не допускаются к производству лекарственных средств.
- Одежда сотрудника, входящего в производственные помещения, должна соответствовать назначению этих помещений.
- В производственных и складских зонах запрещаются курение, прием пищи или питье, же- вание резинки, а также хранение пищевых продуктов, напитков, табачных изделий и личных лекарст- венных средств. Как правило, не допускается любая деятельность, нарушающая установленные пра- вила гигиены в производственных помещениях или других местах, которая может оказать отрица- тельное влияние на качество продукции.
- Непосредственный контакт рук операторов с открытой продукцией или любыми деталями оборудования, контактирующими с продукцией, не допускается.
- Персонал должен пройти инструктаж по правилам мытья рук.
- Специальные требования, относящиеся к производству отдельных видов продукции, на- пример, стерильных препаратов, приведены в приложениях к настоящему стандарту.
Консультанты
-
- Консультанты должны иметь необходимое образование, подготовку и опыт или любую ком- бинацию их, чтобы оказывать консультации, для которых они приглашены. Следует сохранять докумен- тацию с указанием имен, адресов, квалификации и видов оказанных этими консультантами услуг.
3.Помещения и оборудование
Основные принципы
Место расположения, проект, строительство, монтаж, оснащение и обслуживание помещений и оборудования должны соответствовать характеру выполняемых работ. Планировочные решения по- мещений и конструкция оборудования должны минимизировать риск ошибок, предусматривать про- ведение эффективной уборки и обслуживания с целью предотвращения перекрестного загрязнения, появления пыли или грязи и, в общем случае, любого фактора, ухудшающего качество продукции.
Помещения
Общие положения
-
- Помещения должны быть расположены так, чтобы, в сочетании с другими факторами, риск загрязнения материалов и продукции был минимальным.
- Текущее содержание помещение должно выполняться тщательно, при этом техническое обслуживание и ремонт не должны оказывать отрицательного влияния на качество продукции. Уборка и, где требуется дезинфекция, помещений должны проводиться в соответствии с подробными пись- менными инструкциями.
- Освещение, температурный режим, влажность воздуха и вентиляция должны соответство- вать назначению помещения и не оказывать прямого или косвенного отрицательного влияния на ле- карственные средства во время их изготовления и хранения и на работу оборудования.
- При проектировании и оснащении помещений должна быть обеспечена максимальная за- щита от проникания в них насекомых или животных.
- В помещения не должны допускаться лица, не имеющие права доступа в них. Производст- венные, складские помещения и помещения контроля качества не должны использоваться для сквоз- ного прохода персонала, не работающего в них.
Производственная зона
-
- Проектом и эксплуатацией производственных помещений должно быть предусмотрено не- допущение перекрестных загрязнений любой продукции. Меры по предотвращению перекрестных загрязнений должны соответствовать степени риска. Для оценки и предотвращения риска следует использовать методы анализа рисков.
В зависимости от степени риска могут потребоваться отдельные помещения или оборудование для производственных операций и/или операций по упаковке, чтобы предотвратить риск, создавае- мый некоторыми лекарственными средствами.
Отдельные помещения и оборудование для производства лекарственных средств, представ- ляющих риск, когда:
- невозможно контролировать риск эксплуатационными и/или техническими средствами;
- отсутствуют научные данные по токсикологической оценке, позволяющие оценивать риск (напри- мер, по аллергенной реакции на высоко сенсибилизирующие материалы, такие как бета лактамы) или
- предельно допустимые значения остатков, полученные при токсикологической оценке, не мо- гут быть удовлетворительно проверены аттестованными аналитическими методами.
Дальнейшие указания содержатся в разделе 5 и приложениях 2; 3; 4; 5 и 6.
-
- Планировочные решения помещений, как правило, должны соответствовать логической по- следовательности производственных операций и обеспечивать выполнение требований к чистоте.
- Планировочные решения рабочих зон и зон хранения внутри производства должны обеспе- чивать последовательное и логичное размещение оборудования и материалов, сводить к минимуму риск перепутывания различных лекарственных средств или их компонентов, перекрестного загрязне- ния и ошибочного выполнения или пропуска любых операций по производству или контролю.
- Если исходные и первичные упаковочные материалы, промежуточные или нерасфасован- ные продукты подвергаются воздействию окружающей среды, внутренние поверхности помещений (стены, пол и потолок) должны быть гладкими, не иметь открытых соединений и трещин, не выделять частиц и должны обеспечивать возможность беспрепятственной и эффективной уборки, а также, при необходимости, дезинфекции.
- Конструкция и размещение труб, осветительных приборов, оборудования вентиляции и т. п. не должны иметь мест, труднодоступных для очистки. По возможности их обслуживание должно осуществляться с внешней стороны производственных помещений.
- Сливы для стоков (в канализацию) должны иметь необходимые размеры и быть оборудо- ваны устройствами, предотвращающими обратный поток. Следует избегать применения открытых желобов. При необходимости они должны быть неглубокими для удобства очистки и дезинфекции.
- В производственных зонах, в зависимости от выпускаемой продукции, выполняемых опе- раций и наружной среды следует предусматривать эффективную систему вентиляции с обеспечени- ем требуемой температуры и, при необходимости, влажности воздуха и его фильтрации.
- Исходные материалы взвешивают, как правило, в специально оборудованных для этого помещениях.
- Если выполнение работы сопровождается выделением пыли (например, при отборе проб, взвешивании, смешении, производственных операциях и упаковке сухих продуктов), то необходимо предусмотреть меры по предотвращению перекрестного загрязнения и проведению очистки.
- При проектировании (в т. ч. разработке планировочных решений) помещений для упаковки лекарственных средств следует предусматривать специальные меры, предотвращающие перепуты- вание или перекрестное загрязнение материалов и продукции.
- Производственные помещения должны быть хорошо освещены, особенно в местах прове- дения визуального контроля.
- Внутрипроизводственный контроль может проводиться в зоне производства, если это не создает помех для технологического процесса.
Зоны складирования
-
- Зоны складирования должны иметь достаточную вместимость для обеспечения надлежа- щего хранения различных категорий материалов и продукции (исходных и упаковочных материалов; промежуточной, нерасфасованной и готовой продукции; продукции, находящейся в карантине; раз- решенной для выпуска, отклоненной, возвращенной или отозванной продукции).
- При проектировании и организации зон складирования следует предусматривать надле- жащие условия хранения. Зоны складирования должны быть чистыми и сухими, в них должен быть обеспечен требуемый температурный режим. При необходимости следует обеспечивать специаль- ные условия хранения (например, температура и влажность воздуха) и их контроль.
- В зонах приема и выдачи материалов и продукции должна быть обеспечена их защита от неблагоприятного влияния окружающей среды. Проект зоны приемки должен предусматривать очист- ку упаковок с поступающими материалами перед их складированием.
- Если режим карантина обеспечивается хранением продукции в раздельных зонах, то эти зоны должны быть четко обозначены. Доступ в них должен быть разрешен только лицам, имеющим
на это право. Любая другая система, заменяющая физическое разделение, должна обеспечивать эк- вивалентную безопасность.
-
- Отбор проб исходных материалов, как правило, следует выполнять в отдельной зоне. При отборе проб в складской зоне должны быть приняты меры, предотвращающие прямое или перекре- стное загрязнение.
- Отклоненные, отозванные или возвращенные материалы и продукцию следует хранить в изолированных зонах.
- Сильнодействующие вещества и препараты должны храниться в зонах с соблюдением мер безопасности и сохранности.
- Хранение печатных материалов ввиду их ключевой роли в подтверждении идентичности лекарственных средств должно быть организовано с соблюдением мер безопасности и сохранности.
Зоны контроля качества
-
- Как правило, лаборатории контроля качества должны быть отделены от производственных помещений. Это особенно важно для лабораторий контроля биологических, микробиологических пре- паратов или радиоизотопов, которые также должны быть отделены друг от друга.
- Проект контрольных лабораторий должен соответствовать требованиям к выполняемым в них операциям. Площадь лабораторий должна быть достаточной для исключения перепутывания и перекрестного загрязнения, а также для хранения образцов и документации.
- Для размещения чувствительных приборов, нуждающихся в защите от вибрации, электро- магнитных полей, повышенной влажности воздуха или других внешних факторов, могут быть преду- смотрены отдельные помещения.
- Особые требования предъявляются к лабораториям, в которых проводятся работы с об- разцами специфических веществ, например, биологическими или радиоактивными материалами.
Вспомогательные зоны
-
- Комнаты отдыха и приема пищи должны быть отделены от других зон.
- Помещения для переодевания, умывания и туалеты должны иметь удобный доступ и соот- ветствовать численности персонала. Не допускается выход из туалетов непосредственно в производ- ственные или складские зоны.
- Участки по ремонту и техническому обслуживанию должны быть, по возможности, отделе- ны от производственных помещений. При необходимости хранения запасных частей и инструментов в зоне производства должны быть предусмотрены специальные помещения или шкафы.
- Помещения для содержания животных должны быть изолированы от остальных зон, иметь отдельный вход (для животных) и отдельные системы вентиляции.
Оборудование
-
- Конструкция, монтаж и порядок технического обслуживания оборудования должны соот- ветствовать его назначению.
- Работы по ремонту и техническому обслуживанию оборудования не должны оказывать от- рицательного влияния на качество продукции.
- Конструкция производственного оборудования должна обеспечивать удобство и возмож- ность его очистки. Операции по очистке оборудования должны выполняться в соответствии с подроб- ными письменными инструкциями. Оборудование следует содержать в сухом и чистом состоянии.
- Инвентарь и материалы для мытья и очистки не должны быть источниками загрязнения, что следует учитывать при их выборе и использовании.
- Оборудование должно быть установлено так, чтобы, по возможности, исключить риск за- грязнения или выполнения ошибочных действий.
- Технологическое оборудование не должно представлять собой какую-либо опасность для продукции. Части технологического оборудования, контактирующие с продукцией, не должны всту- пать с ней в химическую реакцию, выделять или абсорбировать вещества, оказывающие влияние на качество продукции и, таким образом, представлять опасность.
- Пределы измерения и точность весов и другого измерительного оборудования должна со- ответствовать производственным и контрольным операциям, в которых они используются.
- Периодичность калибровки (поверки) измерительных, регистрирующих, контрольных при- боров и весов должна соответствовать требованиям инструкций и методик на эти приборы. Результа- ты калибровки (поверки) должны быть оформлены документально.
- Стационарные трубопроводы должны иметь маркировку с указанием проходящих по ним веществ и, если требуется, направления потока.
- Трубопроводы для дистиллированной, деионизованной и других необходимых видов воды*
* К таким видам воды относятся вода очищенная, вода для инъекций и другие виды в соответствии с фар- макопейными требованиями (прим. разработчика стандарта).
следует обрабатывать в соответствии с инструкциями, в которых указаны пределы микробного за- грязнения и принимаемые меры в случае их превышения.
-
- Неисправное оборудование следует удалять из зоны производства и контроля качества или, по крайней мере, обозначать соответствующим образом.
4. Документация
Общие положения
Правильно разработанная документация является важной частью системы обеспечения каче- ства и основой работы в соответствии с правилами GMP. Различные виды документов и носителей информации должны быть полностью определены в системе обеспечения качества. Документация может быть представлена в различной форме, включая бумажные, электронные носители или фото- графии. Основной целью системы документации должно быть, установление, управление, контроль всех видов деятельности, которые прямо или косвенно влияют на все стороны качества лекарствен- ных средств. Система обеспечения качества должна включать достаточно подробные инструкции, чтобы достичь одинакового понимания требований, удовлетворительного протоколирования выпол- нения различных процессов и оценки любых наблюдений, что позволяет показать в последующем выполнение требований.
Существует два основных вида документов, используемых в обеспечении и документальном подтверждении выполнения требований GMP: инструкции (указания, требования) и протоколы (отче- ты). Для каждого вида документов должны выполняться требования ведения документации.
Документация должна быть точной, полной, доступной и четкой. Выполнение этих условий сле- дует контролировать. Инструкции должны не содержать ошибок и быть в письменной форме. Термин
«письменный» означает «записанный» или находящийся на носителе информации, с которого дан- ные могут быть представлены в читаемой человеком форме.
Виды документов, требуемые правилами GMP
Сайт мастер файл, информация о предприятии (Site Master File): Документ, содержащий описание данных о предприятии, имеющих отношение к GMP.
Документы, содержащие указания и требования:
спецификация (specification): Документ, содержащий подробные требования к материалам и продуктам, используемым или получаемым при производстве; является основой для оценки качества; промышленный регламент, технологическая инструкция и инструкции по упаковке и кон- тролю (manufacturing formulae, processing, packaging and testing instructions): Документы, определяю- щие все используемые исходные материалы, оборудование и компьютерные системы (если преду- смотрены) и содержащие требования ко всем инструкциям по производству, упаковке, отбору проб и контролю. Должны быть заданы требования к внутрипроизводственному контролю и аналитическим
технологиям в процессе, если предусмотрено, совместно с критериями приемлемости;
инструкция, методика (procedure, standard operating procedures, SOP): Документ, содержащий указания по выполнению отдельных видов операций**;
техническое соглашение (technical agreement): документ, отражающий соглашение между за- казчиком и исполнителем при работе по контрактам;
Протоколы и отчеты:
Протокол (Record): Документ, подтверждающий выполнение действия и показывающий соот- ветствие инструкциям, например, работы, события, исследований, а для протоколов серии — историю каждой серии продукции, включая ее реализацию. Протокол включает первичные данные, которые должны использоваться для получения других данных. Субъекты, работающие в поднадзорной сфере и оформляющие протоколы в электронной форме, должны определить, какие документы следует ис- пользовать в качестве первичных.
Паспорт анализа (Certificate of Analysis): документ, содержащий итог результатов контроля об- разцов продукции или материалов1 совместно с оценкой соответствия спецификации.
Отчет (report): Документ о выполнении какой-либо работы, проекта или исследований совмест- но с их результатами, выводами и рекомендациями.
** В оригинале правил GMP EC используется термин «protocol»: документ, содержащих указания по выполне- нию отдельных операций и оформлению их выполнения. В английском тексте тот термин является синонимом термина «инструкция». В русском языке термин «протокол» имеет другое значение (прим. разработчика стандарта).
1 Аналитический отчет может быть основан целиком или частично на данных, полученных в реальном времени (выводы или отчеты об отклонениях) при использовании технологии анализа данных в процессе (РАТ), параметрах или
при измерениях согласно регистрационному досье.
Разработка и контроль документации
-
- Следует разработать все типы документов и обеспечить их выполнение. Установленные требования распространяются на все виды документов на носителях информации. Следует понимать работу сложных систем, оформить их документально, аттестовать (испытать) и организовать необхо- димый контроль. Многие документы (инструкции/или протоколы) могут существовать в гибридной форме, т.е. некоторые элементы могут быть электронной, а другие в бумажной форме. Следует оп- ределить взаимосвязь и методы контроля оригиналов документов, официальных копий, протоколов и порядка обращения данных как для гибридных, так и для однородных систем. Следует ввести поря- док контроля документов в электронной форме, таких как шаблоны, формы и оригиналы документов. Следует ввести порядок контроля для обеспечения целостности протоколов в течение всего срока хранения.
- Следует тщательно выполнять разработку, оформления, пересмотра, и выдачи документов. Они должны соответствовать требованиям, установленным в спецификации на продукцию, его реги- страционном досье, а также в других документах, подаваемых для получения лицензии на производ- ство. Снятие копий с оригиналов документов должно исключать появление ошибок.
- Документы, содержащие указания, должны быть подписаны и утверждены лицами, имею- щими право подписи с указанием даты. Документы не должны допускать двусмысленного толкования и должны иметь собственные обозначения. Следует указывать срок действия документа.
4.4. Документы, содержащие указания, должны иметь логичную структуру, обеспечивающую простоту его проверки. Язык и стиль документов должен соответствовать их назначению. Инструкции и методики должны быть написаны в императивном, указывающем стиле.
-
- Документы, входящие в систему качества, следует регулярно пересматривать и актуализи- ровать.
- Не рекомендуется оформление документов в рукописном виде, но при необходимости вно- сить данные вручную следует предусматривать достаточно свободного места.
Правила ведения документации
-
- Вносить данные в документ от руки следует четким, разборчивым почерком так, чтобы их нельзя было удалить.
- Протоколы следует оформлять одновременно с выполнением соответствующих действий таким образом, чтобы можно было проследить все основные операции при производстве лекарствен- ных средств.
- При внесении изменений в документы следует проставлять дату внесения изменения и под- пись лица, внесшего это изменение. Внесенные изменения не должны создавать затруднений для восприятия исходного текста. При необходимости следует указать причину внесения изменений.
Хранение документов
-
- Следует четко указывать, к какой производственной деятельности относится данный про- токол и где он хранится. Сохранность и целостность протоколов должны быть обеспечены в течение всего срока хранения и, при необходимости, аттестованы.
- Специальные требования предъявляются к документации на серию продукции, которую следует хранить не менее одного года со дня окончания срока годности серии или пяти лет после вы- пуска серии, в зависимости от того, какой из сроков заканчивается позже. Для лекарственных средств, предназначенных для исследований, документация на серию следует хранить не менее пяти лет после окончания или формального прекращения последнего клинического исследования, в кото- ром эта серия использовалась. Другие требования к хранению документации могут содержаться в законодательстве, относящемуся к специфическим видам продукции (например, к новейшим терапев- тическим лекарственным средствам) и указываться более длительные сроки хранения отдельных до- кументов.
- Следует установить сроки хранения всех видов документации в зависимости от деятель- ности, к которой она относится. Критическую документацию, включая первичные данные (например, относящуюся к аттестации или стабильности)), которая выполнена в соответствии со свидетельством о регистрации, следует хранит в течение всего срока действия этого свидетельства. Может быть до- пущено изъятие некоторых документов (например, первичных данных из отчетов по аттестации или отчетов об исследовании стабильности), если получены новые данные в полном объеме. Это следует обосновать с учетом требований к хранению документации на серию продукции; например, первич- ные данные для процессов аттестации следует хранить не менее срока хранения протоколов всех серий, которые выпущены га основе этих процессов аттестации.
Ниже приводятся примеры требуемых документов. Система обеспечения качества должна включать описание всех документов, требуемых для обеспечения качества и безопасности пациента.
Спецификации
-
- Следует разработать и утвердить спецификации на исходные, упаковочные материалы и готовую продукцию с указанием даты.
Спецификации на исходные и упаковочные материалы
4.11 Спецификации на исходные материалы, первичную упаковку или печатные материалы должны включать в себя следующие данные, где применимо:
- описание материалов, в т. ч.:
- наименование и внутризаводской код;
- ссылку на фармакопейную статью или другую нормативную документацию (при ее наличии);
- наименование утвержденных поставщиков и, по возможности, первичного производителя ма- териалов;
- образец печатных материалов;
- методики отбора проб и проведения испытаний или ссылки на соответствующие методики;
- количественные и качественные характеристики с указанием допустимых предельных значений;
- условия хранения и меры предосторожности;
t) максимальный срок хранения до повторного контроля.
Спецификации на промежуточную и нерасфасованную продукцию
-
- Спецификации на промежуточную и нерасфасованную готовую продукцию должны быть в наличии для критических стадий или при ее приобретении или отгрузке. Эти спецификации должны быть аналогичны спецификациям на исходные материалы или готовую продукцию в зависимости от их вида.
Спецификации на готовую продукцию
-
- Спецификации на готовую продукцию должны включать в себя следующие данные:
- наименование продукции и код (при необходимости);
- состав лекарственного средства или ссылку на соответствующий документ;
- описание лекарственной формы и данные об упаковке;
- методики отбора проб и проведения испытаний (или ссылки на них);
- количественные и качественные характеристики с указанием допустимых предельных значений;
- условия хранения и особые меры предосторожности при обращении с лекарственным сред- ством (при необходимости);
- срок годности.
Промышленный регламент и технологические инструкции
Для каждого вида продукции и размера серии должны быть разработаны и утверждены про- мышленный регламент и технологические инструкции
-
- Промышленный регламент должен включать в себя:
- наименование продукции и код в соответствии со спецификацией;
- описание лекарственной формы, ее дозировки и размер серии;
- перечень всех исходных материалов с точным наименованием и указанием их количества; указание на все вещества, которые могут преобразовываться в ходе технологического процесса;
- ожидаемый выход готовой продукции с указанием допустимых пределов и выход промежу- точных продуктов (при необходимости).
-
- Технологические инструкции включают в себя:
- данные о месте нахождения производства и основном оборудовании;
- инструкции по подготовке основного (критического) оборудования (например, по очистке, сборке, калибровке (поверке), стерилизации) или ссылки на них;
- указание на проверку оборудования и рабочих мест на предмет отсутствия остатков предше- ствующей продукции, документов или материалов, не требующихся в данном процессе, проверку чистоты и готовности к работе;
- подробное постадийное описание технологического процесса (например, по контролю мате- риалов, предварительной обработке, последовательности внесения материалов, времени перемеши- вания, температуре и т.д.);
- описание всех видов внутрипроизводственного контроля с указанием допустимых пределов;
- условия хранения нерасфасованной продукции (в т. ч. требования к упаковке и маркировке) и специальные условия хранения (при необходимости);
- специальные меры предосторожности.
Инструкции по упаковке
-
- Для каждого вида продукции, размера и вида упаковки должны быть разработаны и утвер- ждены инструкции по упаковке, которые включают в себя или содержат ссылку на:
- наименование продукции, включая номер серии нерасфасованной или готовой продукции;
- описание ее лекарственной формы и дозировки (при необходимости);
- количество продукта в окончательной упаковке, выраженное в единицах измерения (штуках, единицах массы или объема);
- перечень всех упаковочных материалов, в т. ч. количество, размер и тип упаковочного мате- риала с указанием кода или номера в соответствии с их спецификацией;
- образец или копию соответствующего печатного упаковочного материала (при необходимо- сти) и образцы с указанием места нанесения номера серии и срока годности продукта;
- указание на проверку оборудования и рабочих мест на предмет отсутствия остатков предше- ствующей продукции, документов или материалов, не требующихся в данных операциях по упаковки (очистку линии), проверку чистоты и готовности к работе;
- специальные меры предосторожности, в т. ч. тщательную проверку оборудования и зоны упаковки, гарантирующие чистоту упаковочной линии перед началом работы;
- описание процесса упаковки со всеми основными вспомогательными операциями и исполь- зуемым оборудованием;
- подробное описание проведения внутрипроизводственного контроля, в т. ч. порядок отбора проб и указание допустимых пределов.
Протоколы на серию продукции
-
- На каждую серию продукции должен быть составлен и храниться протокол (протокол на серию продукции). Он должен основываться на соответствующих требованиях действующих про- мышленных регламентов и технологических инструкций. Протокол на серию продукции должен вклю- чать в себя следующие данные:
- наименование продукта и номер серии;
- дату и время начала и окончания основных промежуточных этапов и завершения технологи- ческого процесса;
- фамилию и инициалы сотрудника, ответственного за выполнение каждой производственной стадии, фамилию и инициалы лица, проверяющего выполнение (где это применимо)*;
- номер серии и/или номер анализа, а также фактическое количество взвешенных исходных материалов, в т. ч. номер серии и количество добавленных регенерированных или переработанных материалов;
- основные технологические операции или действия, а также основное оборудование;
- протоколы внутрипроизводственного контроля с указанием исполнителей и полученных ре- зультатов;
- выход продукции, полученной на основных производственных стадиях;
- подробное описание любых отклонений от промышленного регламента и технологических инструкций, подписанное ответственным лицом;
- утверждение протокола лицом, ответственным за производство.
Примечание – Если предусмотрены непрерывный контроль и регулирование аттестованного процес- са, то формируемые автоматически протоколы могут быть сокращены до сводных данных о соответствии и от- клонениях от спецификаций.
Протоколы на упаковку серии продукции
-
- Для каждой серии продукции должен быть составлен протокол на упаковку, который сле- дует хранить в установленном порядке. Протокол должен соответствовать требованиям действующих инструкций по упаковке продукции. Протокол на упаковку серии продукции должен включать в себя следующие данные:
- наименование продукта и номер серии;
- дату(ы) и время операций по упаковке;
- фамилии и инициалы упаковщиков, выполнивших различные стадии упаковки, фамилию и инициалы лица, проверившего выполнение этих операций, где это применимо;
- протоколы проверки соответствия упаковки требованиям инструкций по упаковке, в т. ч. ре- зультаты внутрипроизводственного контроля;
- подробные данные о выполнении операций по упаковке, в т. ч. ссылки на используемое обо- рудование и упаковочные линии;
- образцы печатных материалов, в т. ч. образцы с обозначением номера серии, срока годности и любого лишних печатных материалах, везде, где это возможно;
- подробное описание любых отклонений от инструкций по упаковке и необычных событий и подписанное разрешение на каждое отклонение;
- количество и наименования или коды вех выданных, использованных, уничтоженных или возвращенных на склад печатных материалов и нерасфасованной продукции и количество получен-
- В предыдущей редакции правил GMP EC в данном пункте в качестве примера операций, требующих кон- троля вторым лицом, было указано взвешивание (прим. разработчика стандарта).
ной готовой продукции для составления общего баланса. Эта информация может не указываться при наличии строго электронного контроля процесса упаковки;
- утверждение протокола лицом, ответственным за упаковку.
Инструкции и протоколы
Приемка
-
- Приемку каждой серии каждого вида поставляемых исходных материалов (включая не- расфасованную, промежуточную и готовую продукцию), первичных и вторичных упаковочных мате- риалов и печатных материалов следует проводить в соответствии с письменной инструкцией. По ре- зультатам приемки должен быть оформлен протокол.
- Протоколы приемки должны включать в себя следующие данные:
- наименование материала по накладной и обозначение на упаковке;
- внутризаводское наименование или код материала (если они отличаются от указанных в пе- речислении а));
- дату приемки;
- наименования поставщика и производителя;
- номер серии производителя или ссылочный номер;
- общее количество полученных материалов и число единиц упаковки;
- номер серии, присвоенный после приемки;
- замечания, относящиеся к предмету (например, о состоянии упаковки)*.
-
- На предприятии должны быть письменные инструкции по внутризаводской маркировке, ка- рантину и хранению исходных, упаковочных и других материалов, исходя их потребности.
Отбор проб
-
- На предприятии должны быть письменные инструкции по отбору проб должны быть указа- ны методы отбора проб и оборудование, количество отбираемых материалов и меры предосторожно- сти, исключающие загрязнение или ухудшение качества продукции.
Проведение испытаний
-
- На предприятии должны быть письменные методики испытаний материалов и продукции на различных этапах производства с указанием используемых методов и оборудования. Результаты испытаний оформляются в виде протокола.
Прочее
-
- На предприятии должны быть письменные инструкции по выпуску и отклонению материа- лов и продукции, в особенности регламентирующие выдачу уполномоченным лицом разрешений на выпуск продукции для реализации. Должна действовать система, указывающая на специфические наблюдения или любые изменения критических данных.
- Документацию на реализацию каждой серии продукции следует хранить для обеспечения (при необходимости) возможности ее оперативного отзыва в случае необходимости.
- На предприятии должны быть письменные инструкции, протоколы и отчеты на следующее:
- аттестацию (испытания процессов, оборудования и систем;
- монтаж и калибровку (поверку) оборудования;
- передачу технологий;
- техническое обслуживание, очистку и дезинфекцию;
- обучение персонала по GMP и техническим предметам, одежде, соблюдение правил личной гигиены и порядка контроля эффективности обучения, с листами с подписями;
- контроль окружающей среды;
- борьбу с паразитами, насекомыми и другими животными;
- рекламации;
- отзывы продукции;
- возвраты продукции;
- контроль изменений;
- расследование отклонений и несоответствий;
- результаты внутренних аудитов и аудитов на соответствие GMP;
- результаты аудитов поставщиков.
-
- На предприятии должны быть по эксплуатации основного производственного и контрольно- аналитического оборудования.
- Для основного или критического производственного, контроль-аналитического оборудования и производственных зон следует вести журналы с перечислением в хронологической последовательно- сти выполняемых в них работ, калибровке (поверке), обслуживанию, очистке или ремонту, в т. ч. с ука-
- в предыдущей редакции правил GMP EC в данном пункте, подпункт h), в качестве примера замечаний ука- зывалось состояние упаковки (прим. разработчика стандарта).
занием дат, фамилий и инициалов, а также подписей лиц, проводивших эти операции.
-
- Следует вести ведомость всех документов системы обеспечения качества.
5. Производство
Основные принципы
Для получения продукции требуемого качества технологические операции следует выполнять согласно промышленному регламенту и соответствующим инструкциям, требованиям настоящего стандарта, нормативных документов и требованиям, установленным при регистрации лекарственного средства.
Общие положения
-
- Производственный процесс и его контроль должны осуществляться квалифицированным пер- соналом.
- Все операции с материалами и продукцией (например, приемка, карантин, отбор проб, хране- ние, маркировка, подготовка, приготовление, упаковка и отгрузка) должны выполняться согласно пись- менным инструкциям или методикам и, при необходимости, протоколироваться.
- Все поступающие материалы должны быть проверены на соответствие заказу. Тару и упаков- ку следует очищать и маркировать.
- Факты повреждения тары и упаковки, которые могут оказать отрицательное влияние на каче- ство материалов, следует расследовать и протоколировать с последующим сообщением об этом в от- дел контроля качества.
- Поступающие материалы и произведенная готовая продукция должны немедленно поме- щаться в карантин, действующий по принципу раздельного хранения или за счет организационных мер, и содержаться в нем до получения разрешения на использование или отгрузку.
- Приемку промежуточной и нерасфасованной продукции проводят в соответствии с правила- ми, действующими для исходных материалов.
- Все материалы и продукцию следует хранить в соответствующих условиях, установленных производителем, в порядке, обеспечивающем разделение серий продукции и ее оборот на складе.
- Для гарантии отсутствия отклонений за допустимые пределы следует обеспечить контроль вы- хода (выпуска) продукции и количественное сопоставление его с данными промышленного регламента.
- Не допускается одновременное или последовательное проведение операций с различными продуктами в одном и том же помещении при отсутствии защиты от риска перепутывания или перекре- стного загрязнения.
- Продукция и материалы должны быть защищены от микробного и других видов загрязнений на всех этапах производства.
- При работе с сухими материалами и продуктами необходимо принимать особые меры пре- досторожности по предотвращению образования и распространения пыли, особенно при работе с сильнодействующими и сенсибилизирующими веществами.
- В ходе выполнения технологического процесса на всех материалах, упаковках с нерасфасо- ванной продукцией, основном оборудовании и помещениях должны быть обозначения (маркировка) с указанием производимой продукции или материала, его дозировкой (при необходимости) и номера се- рии. При необходимости следует указывать стадию технологического процесса.
- Обозначения (маркировка) на упаковке, оборудовании или помещениях должны быть четки- ми, однозначными, установленной формы. Кроме применения буквенных обозначений рекомендуется использовать цветовую маркировку, указывающую статус продукции (например, «Карантин», «Приня- то», «Отклонено», «Чистое» и т. п.).
- Следует контролировать правильность соединения трубопроводов и другого оборудования, предназначенного для транспортирования продукции из одной зоны в другую.
- Отклонение от инструкций не допускается. При необходимости письменное разрешение на отклонение от инструкций должно быть получено от компетентных лиц и отдела контроля качества.
- В производственные помещения может входить только персонал, имеющий право доступа
в них.
Предотвращение перекрестного загрязнения при производстве
-
- Как правило, в помещениях и на оборудовании, предназначенных для производства лекар-
ственных средств, не допускается изготовление продукции немедицинского назначения, но при нали- чии обоснования, может быть разрешено, если приняты меры, предотвращающие перекрестные за- грязнения с лекарственными средствами, указанными ниже и в главе 3. Не опускается производство и/или хранение технических ядов, таких как пестициды (кроме используемых в производстве лекарст-
венных средств) гербицидов в зонах для производства и/или хранения лекарственных средств.
-
- Следует исключить возможность загрязнения исходных материалов или продуктов другими материалами или продуктами. В процессе производства риск случайного перекрестного загрязнения возникает при неконтролируемом выделении пыли, газов, испарений, аэрозолей, генные материалов или организмов из активных субстанций, других, чем исходные материалы и продукция в процессах и от остаточных загрязнений на оборудовании и одежде людей. Степень риска зависит от типа загряз- нения и продукта, подверженного загрязнению. Наиболее серьезными считаются перекрестные за- грязнения продукции, вводимой путем инъекции или в течение длительного времени. Однако загряз- нение любой продукции представляет риск для безопасности пациента в зависимости от природы и количества загрязнений.
- Перекрестные загрязнения следует предупреждать на стадии проектирования помещений и оборудования согласно главе 3. При этом следует должным образом разработать технологический процесс и предусмотреть все необходимые технические и организационные меры, включая эффек- тивные и воспроизводимые процессы очистки для предупреждения перекрестных загрязнений.
- Для оценки и контроля риска загрязнений в производстве лекарственных средств следует применять анализ рисков, который включает в себя оценку активности и токсичности. Следует также принять во внимание проектные решения и конструкцию оборудования, порядок эксплуатации, потоки (маршруты движения) персонала и материалов, микробиологические показатели, физико-химические характеристики активных субстанций, характеристики технологического процесса, процессов очистки и возможности аналитических методов по обнаружению остатков при их предельных величинах, ус- тановленных при анализе продукции. Результаты анализа рисков служат основой для принятия ре- шения о необходимости и масштаба специализации помещений и оборудования для производства продукта или группы продуктов. К этому может относиться специализация частей оборудования, вступающих в контакт с продуктом, или специализация всего производства. Может быть предусмот- рено выполнение производственных операций в разделенных (выделенных) зонах в пределах одного многономенклатурного производства, при наличии обоснования.
- На основе анализа рисков следует принять решения о технических и организационных ме- рах по предупреждению перекрестных загрязнений. Эти меры могут включать, но не ограничиваться следующим:
Технические меры
- Специализированные производственные помещения и оборудование;
- Выделенные производственные зоны с отдельным технологическим оборудование и от- дельными системами отопления, вентиляции и кондиционирования воздуха. Может оказаться целе- сообразным изолировать отдельные системы от используемых в других зонах;
- Проектирование производственных процессов, помещений и оборудования с условием све- дения к минимуму возможности перекрестных загрязнений при производстве, техническом обслужи- вании и очистке;
- Использование «закрытых систем» в производстве и передаче материалов (продукции) ме- жду единицами оборудования;
- Использование систем с физическими барьерами, включая изоляторы;
- Удаление пыли из зон вблизи источника загрязнений за счет местных вытяжек;
- Специализация оборудования, частей, контактирующих с продуктом или специализация от- дельных трудно поддающихся очистке частей (например, фильтров), специализация инструментов для обслуживания;
- Использование одноразовых технологий;
- Использование оборудования, конструкция которого предусматривает легкость очистки;
- Организация воздушных шлюзов и каскадов с перепадам давления для ограничения распро- странения загрязнений за пределы определенных зон;
- Сведение к минимуму риска загрязнений, вызываемых рециркуляций воздуха, поступлением не подготовленного или недостаточно подготовленного воздуха;
- Использование автоматических систем очистки на месте с подтвержденной эффективностью;
- В обычных зонах для мойки разделение зон мойки, сушки и хранения оборудования.
Организационные меры
- Специализация целых производств (помещений и оборудования) или выделение зон с раз- делением циклов производства во времени уборкой (очисткой) процессы которой аттестованы;
- Хранение специальной защитной одежды в пределах зон производства продукции с высоким риском перекрестного загрязнения;
- Проверка чистоты после каждого цикла производства, которая должна подтверждать эф- фективность принятых на основе анализа рисков мер по защите продукции с повышенным риском;
- В зависимости от риска загрязнений проверка чистоты поверхностей, не контактирующих с продуктом после их обработки, текущий контроль чистоты воздуха в производственных зонах и/или примыкающих зонах для подтверждения эффективности мер против аэрозольных загрязнений или
механического переноса загрязнений;
- Специальные меры по удалению отходов, загрязненной воды после мойки загрязненной одежды;
- Регистрация проливов, случайных событий или отклонений от документации;
- Разработка процессов очистки для помещений и оборудования так, чтобы сами эти процес- сы не приводили к перекрестным загрязнениям;
- Разработка форм подробных протоколов процессов очистки для подтверждения заверше- ния очистки в соответствии с утвержденными инструкциями и использование статусных этикеток, обозначающих проведение очистки, а оборудовании и в производственных зонах;
- Использование общих зон для мытья циклами;
- Контроль за поведением персонала для подтверждения эффективности обучения и соответ- ствия инструкциям.
-
- Следует периодически проверять эффективность мер по предотвращению перекрестного загрязнения в соответствии с утвержденными инструкциями.
Аттестация (испытания)
-
- Аттестация (испытания) направлена на повышение эффективности работы и проводится в соответствии с утвержденными методиками. Ее результаты должны быть оформлены документально.
- При утверждении нового промышленного регламента или метода производства следует проверять их пригодность для серийного производства. Должно быть подтверждено, что данный про- цесс, используемые материалы и оборудование позволяют постоянно производить продукцию тре- буемого качества.
- При существенных изменениях технологии, в т. ч. любых изменениях оборудования или материалов, способных влиять на качество продукции или воспроизводимость процесса, следует проводить аттестацию (испытания) соответствующих процессов.
- Для постоянного подтверждения достижения требуемых результатов следует периодиче- ски проводить повторную аттестацию (испытания) технологических процессов и методик.
Исходные материалы
-
- Выбор, аттестация, утверждение и контроль поставщиков исходных материалов, так же как закупка и приемка материалов должны оформляться документально и входить составной частью в фармацевтическую систему качества. Степень контроля должна соответствовать риску, связанному с конкретными материалами, учитывая их происхождение, процесс производства, сложность цепи по- ставки и конечное использование, для которого материал вводится в лекарственное средство. Сле- дует учитывать дополнительные данные о каждом поставщике и утверждении материалов. Зани- мающийся этим персонал должен иметь текущую информацию о поставщиках, цепи поставки и свя- занных с ними рисках. Исходные материалы следует закупать непосредственно у производителя, где это возможно.
- Требования к качеству исходных материалов, установленные производителем* должны быть рассмотрены и согласованы поставщиком. Необходимые вопросы, связанные с производством и контролем исходных материалов, в т. ч. работа с ними, маркировка, упаковка, и реализация, порядок предъявления рекламаций, отзывов и отклонений, должны быть оформлены документально в согла- шении о качестве или в спецификации.
- Для утверждения поставщиков активных субстанций и вспомогательных веществ и их кон- троля необходимо выполнить следующие требования:
Активные субстанции
Цепь поставки должна быть прослеживаемой. Следует оценить и периодически проверять рис- ки в последовательности от активной субстанции до готового лекарственного средства. Следует при- нять необходимые меры по снижению рисков для активных субстанций.
Следует вести и хранить документацию о цепи поставки и ее прослеживаемости для каждой ак- тивной субстанции (включая исходные материалы для субстанции).
Следует проводить аудит производителей и поставщиков активных субстанций для подтвер- ждения их соответствия требованиям правилам производства лекарственных средств (GMP) и прави- лам обращения лекарственных средств в сетях оптовой торговли (GDP). Держатель свидетельства о регистрации должен проверять это соответствие сам или с привлечением субъекта, действующего по его поручению согласно контракту. Для ветеринарных лекарственных средств аудит следует прово- дить на основе анализа рисков.
Аудит должен быть достаточной продолжительности и объема, чтобы дать полную и ясную оценку соответствия требованиям GMP. При его проведении следует оценить возможность перекре-
- лекарственного средства (прим. разработчика стандарта).
стного загрязнения одних материалов другими на данной площадке. Отчет об аудите должен полно- стью отражать, что было сделано и обнаружено во время аудита с четким указанием всех отклоне- ний. Следует предпринять все предупреждающие и корректирующие действия.
Срои следующих аудитов следует определять на основе анализа рисков для процесса исходя из поддержания постоянного соответствия утвержденной цепи поставок установленным требованиям.
Вспомогательные вещества
Следует контролировать вспомогательные вещества и их производителей на основе результа- тов формализованного анализа рисков*.
-
- В каждой поставке исходных материалов следует проверять целостность тары, упаковки и средств контроля вскрытия, при необходимости, на соответствие накладной, заказу, маркировке по- ставщика и утвержденной спецификации производителя и поставщика, согласованной производите- лем лекарственного средства. Результаты контроля каждой приемки следует оформлять докумен- тально.
- Если поступившие материалы состоят из нескольких серий, то каждую серию следует рас- сматривать как независимую в отношении отбора проб, проведения испытаний и получения разреше- ния на использование.
- Исходные материалы, находящиеся на складе, должны быть маркированы соответствую- щим образом (5.13). Маркировка должна содержать, как минимум, следующую информацию:
- обозначение исходного продукта и внутризаводской код;
- номер серии, присвоенный при приемке;
- статус материала (например, «Карантин», «Испытания», «Разрешено», «Отклонено» и т.п.);
- срок годности или дату, после которой необходимо проведение повторного контроля.
Если складское хозяйство полностью оснащено компьютерами, указывать всю эту информацию в маркировке необязательно.
-
- Контроль подлинности содержимого каждой упаковки с исходными материалами регламен- тируется соответствующими инструкциями и методиками. Упаковки с нерасфасованной продукцией, из которых были отобраны пробы, должны иметь соответствующую маркировку (раздел 6).
- При производстве лекарственных средств могут использоваться только исходные мате- риалы с неистекшим сроком годности и допущенные отделом контроля качества.
- Производители готовых лекарственных средств несут ответственность за испытания ис- ходных материалов2 согласно регистрационному досье. Они могут использовать частично или полно- стью результаты испытаний производителей исходных материалов, но обязаны, как минимум, прово- дить тест на идентичность3 каждой серии в соответствии с приложением 8.
- При выполнении испытаний по контракту следует обосновать их допустимость с докумен- тальным оформлением, отразив выполнение следующих требований:
- Особое внимание следует уделить контролю процесса поставки (транспортированию, опто- вой торговле, хранению и отгрузке), в котором должны сохраняться характеристики качества исход- ных материалов и результаты контроля остаются действительными для поставляемого материала;
- Производитель лекарственного средства должен проводить аудиты самостоятельно или с по- мощью третьей стороны с определенными интервалами времени, основываясь на данных о риске у ор- ганизаций, проводящих испытания исходных материалов (включая отбор проб), чтобы подтвердить вы- полнение требований GMP, спецификаций и методов испытаний, приведенных регистрационном досье;
- Паспорт анализа, выполненного производителем или поставщиком исходного материала должен быть подписан лицом, имеющим на то право, достаточную квалификацию и опыт. Подпись удостоверяет, что каждая серия проверена на соответствие согласованной спецификации на продукт, если только это подтверждение на выполняется отдельно;
- Производитель лекарственного средства должен иметь опыт работы с производителем ис- ходного материала (включая работу через поставщика), включая оценку ранее полученных серий и истории соответствия до принятия решения о сокращении испытаний на своем производстве. Следу- ет учитывать любые изменения в производстве и методах испытаний;
- Производитель лекарственного средства должен также выполнять испытания полностью (сам или с помощью согласованной лаборатории, работающей по контракту) через определенные интервалы времени, основываясь на анализе рисков и сравнении с результатами, приведенными в паспорте анализа от производителя, чтобы проверить достоверность этих результатов. Если будут обнаружены расхождения, то нужно провести расследование и принять необходимые меры. В этом случае признание паспорта анализа от производителя материала или поставщика должно быть пре- кращено до выполнения этих мер.
- В оригинале правил GMP EC “Guidelines on the formalised risk assessment for ascertaining the appropriate Good Manufacturing Practice for excipients of medicinal products for human use” (прим. Разработчика стандарта).
2 Аналогичный подход следует применять к упаковочным материалам согласно п. 5.45.
3 Тест на идентичность исходных материалов следует проводить в соответствии с методами и спецификациями в свидетельстве о регистрации (досье).
-
- Исходные материалы должны выдаваться только специально назначенными лицами в со- ответствии с письменной инструкцией, при этом должны взвешиваться только требуемые материалы и выполняться требования к точности взвешивания и отмеривания материалов в чистую и маркиро- ванную тару.
- Следует проводить независимую проверку каждого выданного вещества, его массы и объ- ема с документальным оформлением.
- Материалы, полученные для каждой серии, следует хранить в одном месте, которое долж- но быть четко обозначено.
Промежуточная и нерасфасованная продукция
-
- Перед началом любой технологической операции следует принять меры, гарантирующие чистоту производственной зоны и оборудования, отсутствие любых исходных материалов, продукции, остатков продукции или документации, не относящихся к данному процессу.
- Промежуточную и нерасфасованную продукцию следует хранить в надлежащих условиях.
- Критические процессы должны быть аттестованы (5.23–5.26).
- Выполнение всех необходимых операций по внутрипроизводственному контролю и кон- тролю окружающей среды должно быть оформлено документально.
- Следует регистрировать и расследовать все факты существенного отклонения выхода (ко- личества) продукции от ожидаемого.
Упаковочные материалы
-
- Выбор, аттестация, утверждение и контроль поставщиков первичных упаковочных и печат- ных материалов выполняется аналогично исходным материалам.
- Особое внимание следует уделять печатным материалам. Их следует хранить в безопас- ных условиях, исключающих доступ посторонних лиц. Разрезанные этикетки и другие разрозненные материалы должны храниться и транспортироваться раздельно в закрытой таре, исключающей их перепутывание. Разрешение на использование упаковочных материалов должно выдаваться только специально назначенными лицами в соответствии с утвержденной инструкцией.
- Каждой поставке или серии первичных упаковочных или печатных материалов должен быть присвоен номер или отличительный знак.
- Просроченные или непригодные к использованию первичные упаковочные или печатные материалы должны быть уничтожены с оформлением протокола.
Операции по упаковке
-
- При упаковке продукции должен быть сведен к минимуму риск перекрестного загрязнения, перепутывания или подмены. Не допускается упаковывать продукцию различных видов в непосред- ственной близости друг от друга при отсутствии физического разделения зон.
- Перед началом операций по упаковке следует убедиться в том, что рабочая зона, упако- вочные линии, печатные машины и другое оборудование находятся в чистом состоянии и не содер- жат материалов, продукции или документов, относящихся к предшествующей работе и не исполь- зующихся в текущем процессе. Очистка линии упаковки продукции должна проводиться по специаль- ным инструкциям.
- Наименование и номер серии упаковываемой продукции должны быть указаны на каждой линии или установке.
- При поступлении продукции и упаковочных материалов на участок упаковки следует про- верить их количество, подлинность и соответствие инструкциям по упаковке.
- Первичные упаковочные материалы перед началом операции наполнения должны быть чистыми. Особое внимание следует обратить на удаление любых загрязняющих веществ (осколков стекла, металлических частиц и пр.).
- После наполнения и укупоривания продукции ее маркировку следует выполнять как можно быстрее. Если это невозможно, то следует принять необходимые меры, предотвращающие перепу- тывание продукции или ошибочную маркировку.
- Правильность выполнения любых печатных операций (например, нанесения кодов или срока годности) при упаковке и после нее следует тщательно контролировать и оформлять докумен- тально. Особое внимание следует уделять ручной маркировке, которую следует повторно контроли- ровать через определенные интервалы времени.
- Особые меры предосторожности должны приниматься при использовании разрезанных этикеток и нанесении печати вне линии упаковки. Для предотвращения перепутывания печатного ма- териала рекомендуется использовать этикетки в рулоне вместо разрезанных этикеток.
- Следует контролировать правильность работы электронных считывателей кодов, счетчи- ков этикеток и других подобных устройств.
- Маркировка упаковочных материалов, нанесенная с помощью печати или методом тисне- ния, должна быть отчетливой, устойчивой к воздействию света (выгоранию) и удалению.
- При контроле процесса упаковки продукции на линии следует проверять, как минимум, следующее:
- общий вид упаковки;
- комплектность упаковки;
- правильность использования упаковочных материалов для данной продукции;
- правильность нанесения печатных надписей;
- правильность работы устройств контроля на линии.
Не допускается возврат образцов продукции, отобранных с упаковочной линии, повторно на линию.
-
- Если при упаковке продукции возникли непредвиденные обстоятельства, она может быть возвращена в процесс только после специальной проверки, проведения расследования и с разреше- ния лица, имеющего соответствующие полномочия. Указанные действия должны быть оформлены в виде протоколов, которые следует хранить в установленном порядке.
- При существенном или необычном расхождении, установленном при сопоставлении коли- чества нерасфасованной продукции, печатных упаковочных материалов и количества единиц полу- ченной готовой продукции, следует провести расследование и установить причину этого расхождения до получения разрешения на реализацию данной продукции.
- После завершения операций по упаковке любые оставшиеся упаковочные материалы с нанесенным на них номером серии должны быть уничтожены с последующим составлением протоко- ла. Возврат на склад немаркированных упаковочных материалов следует производить в соответствии с утвержденной инструкцией.
Готовая продукция
-
- До выдачи разрешения на реализацию готовая продукция должна содержаться в каранти- не в условиях, установленных производителем.
- Порядок оценки качества готовой продукции и требования к документации, необходимые для получения разрешения на реализацию, приведены в разделе 6.
- После выдачи разрешения на реализацию готовая продукция должна храниться на складе готовой продукции в условиях, установленных производителем.
Отклоненные, повторно использованные и возвращенные материалы
-
- Отклоненные материалы и продукция должны иметь четкую маркировку и храниться раз- дельно в зонах с ограниченным доступом. Они подлежат возврату поставщику, переработке (если это допустимо) или уничтожению. Любые выполненные действия должны быть документально оформле- ны и утверждены лицами, имеющими соответствующие полномочия.
- Переработка отклоненной продукции допускается в исключительных случаях при условии отсутствия ухудшения качества готовой продукции и выполнения всех требований спецификаций и выполнении всех требований утвержденной инструкции, после оценки связанных с этим рисков. Пе- реработка должна быть оформлена протоколом.
- Повторное использование всей (или части) ранее произведенной серии продукции требуе- мого качества путем объединения ее с другой серией той же продукции на определенном этапе про- изводства допускается только после получения предварительного разрешения, подписанного ответ- ственными лицами. Повторное использование продукции допускается только после оценки возможно- го риска (в т. ч. его влияния на срок годности серии) по утвержденной инструкции с оформлением протокола.
- Необходимость дополнительного контроля готовой продукции, прошедшей переработку, или продукции, в которую была включена ранее изготовленная продукция, определяет отдел контро- ля качества.
- Возвращенная с рынка продукция, над которой был утрачен контроль со стороны произво- дителя, должна быть уничтожена, если не подтверждено соответствие ее качества установленным требованиям. Решение о повторной продаже, перемаркировке или повторном использовании в по- следующей серии может быть принято только после специального анализа, проведенного отделом контроля качества в соответствии с письменной инструкцией. При этом необходимо учитывать харак- тер продукции, ее предысторию и состояние, соблюдение специальных условий хранения и время, прошедшее с даты выпуска. При любых сомнениях в отношении качества продукции не допускается ее повторное использование или повторный выпуск, но допускается ее химическая переработка с це- лью регенерации активных ингредиентов. Все выполняемые действия должны быть оформлены до- кументально.
Прекращение выпуска продукции из-за ограничений в производстве
-
- Производитель должен сообщить держателю регистрационного досье о любых затрудне- ниях в процессе производства, которые могут привести к ограничениям на поставку. Это должно быть сделано своевременно, чтобы держатель регистрационного досье мог известить надзорные органы об ограничениях поставки в соответствии с установленными требованиями.
6. Контроль качества
Основные принципы
Данный раздел следует применять совместно со всеми относящимися к данному предмету пра- вил GMP.
Контроль качества включает в себя отбор проб, спецификации и проведение испытаний и про- верок, организацию работы, документирование и выдачу разрешений на реализацию так, чтобы были проведены все необходимые испытания и проверки и не допустить к использованию или реализации для продажи или снабжения до подтверждения их качества. Деятельность по контролю качества не ограничивается только работой лабораторий, а включает в себя также проведение исследований, проверок и участие в принятии любых решений, касающихся качества продукции. Основополагающим принципом контроля качества является его независимость от производственных подразделений.
Общие положения
-
- На каждом предприятии-изготовителе должен быть отдел контроля качества, независимый от других подразделений. Руководитель этого отдела должен иметь необходимый опыт и квалифика- цию. К отделу контроля качества относятся одна или несколько контрольных лабораторий. Для вы- полнения своих функций отдел должен быть обеспечен всеми необходимыми ресурсами.
- Основные обязанности начальника отдела контроля качества изложены в разделе 2. На от- дел возлагаются также обязанности по разработке, аттестации (испытаниям), внедрению всех инст- рукций (методик) по контролю качества; хранению контрольных и/или архивных образцов материалов и продукции, если предусмотрено; контролю правильности маркировки упаковок с материалами и продукцией; контролю стабильности продукции; участию в расследовании рекламаций, связанных с качеством продукции, и т. п. Все эти функции должны выполняться в соответствии с утвержденными инструкциями с оформлением протоколов (при необходимости).
- При оценке качества готовой продукции следует рассматривать все сопутствующие факто- ры, в т. ч. условия производства, результаты внутрипроизводственного контроля, анализ производст- венной документации (в т. ч. документации на упаковку), соответствие спецификациям на готовую продукцию и состояние окончательной упаковки готовой продукции.
- Сотрудники отдела контроля качества должны иметь доступ в производственные зоны для отбора проб и проведения анализов.
Организация работы контрольных лабораторий
-
- Помещения и оборудование контрольных лабораторий должны соответствовать общим и специальным требованиям, предъявляемым к зонам контроля качества (раздел 3). Как правило, не следует перемещать лабораторное оборудование между зонами с высоким риском случайного пере- крестного загрязнения. Это особенно относится к микробиологическим лабораториям, при организа- ции которых риск перекрестного загрязнения должен быть сведен к минимуму.
- Персонал, помещения и оборудование лабораторий должны соответствовать виду и объему производства. В отдельных случаях допускается использование сторонних лабораторий при условии выполнения ими требований, изложенных в разделе 7, и внесения соответствующих записей в прото- колы контроля качества.
Документация
-
- Документация контрольных лабораторий должна соответствовать требованиям, изложен- ным в разделе 4. К документации по контролю качества относятся:
- Спецификации;
- Методики отбора проб, испытаний, ведения протоколов (в т. ч. аналитические операционные листы и/или лабораторные журналы) и проведению проверок;
- Методики и протоколы проведения калибровки (поверки) приборов, инструкции и журналы технического обслуживания оборудования;
- Инструкции по расследованию случаев выхода за пределы спецификаций или за пределы тенденций;
- Аналитические отчеты и/или паспорта анализов;
- Результаты контроля сред (воздуха, воды и других сред);
- Протоколы аттестации (испытаний) аналитических методов, при необходимости.
Эта информация всегда должна быть готова к представлению в отдел контроля качества.
-
- Документация по контролю качества, относящаяся к протоколам серий продукции, должна храниться в соответствии с требованиями раздела 4).
- Для некоторых данных (например, результатов аналитических испытаний, выходов готовой продукции, параметров окружающей среды и т. п.) целесообразно хранить протоколы в виде, позво- ляющем оценивать тенденции изменения параметров. Случаи выхода за пределы спецификаций или тенденций должны регистрироваться и расследоваться.
- В дополнение к протоколам на серии продукции следует хранить в доступном виде и дру- гую первичную информацию (например, лабораторные журналы и/или протоколы).
Отбор проб
-
- Отбор проб должен проводиться в соответствии с утвержденными инструкциями, вклю- чающими в себя:
- методику отбора проб;
- перечень используемого оборудования;
- количество проб, которое должно быть отобрано;
- инструкции по разделению отобранной пробы на части (при необходимости);
- тип и характеристики тары для отбора проб;
- нанесение маркировки на тару с отобранными пробами;
- специальные меры предосторожности, особенно в отношении стерильных и опасных ве- ществ;
- условия хранения;
- инструкции по очистке и хранению оборудования для отбора проб.
-
- Отобранные контрольные образцы должны представлять собой представительную выбор- ку серии материалов или продукции. Возможен отбор проб, характеризующих критические стадии технологического процесса (например, его начало или окончание). План отбора проб должен быть обоснован с учетом анализа рисков.
- На маркировке тары с отобранными пробами должны быть указаны ее содержимое, даты отбора проб, а также обозначение упаковки, из которой эти пробы были отобраны. При обращении с тарой следует сводить к минимуму риск перепутывания и следует защищать пробы от отрицательно- го влияния условий хранения.
- Руководство по работе с контрольными и архивными образцами приведено в приложении 19.
Проведение испытаний
-
- Аналитические методики должны быть аттестованы*. Если лаборатория использует не атте- стованный первоначально метод, должна проверить его соответствие требованиям (аттестовать). Все испытания, указанные в регистрационном досье, должны быть проведены в соответствии с утвержден- ными методиками.
- Полученные результаты испытаний должны быть оформлены документально. Следует про- верить все данные о параметрах, которые относятся к качеству или являются критическим и оценить тенденцию, убедившись, что они соответствуют друг другу. Все расчеты следует тщательно проверять.
- Проведенные испытания следует оформлять документально с указанием, как минимум:
- Наименования материала или продукции и, при необходимости, лекарственной формы;
- Номера серии и, при необходимости, наименования производителя и/или поставщика;
- Ссылки на соответствующие спецификации и методики испытаний;
- Результатов испытаний, в т. ч. наблюдения, вычисления и ссылки на все паспорта анализов;
- Даты проведения испытаний;
- Фамилий и инициалов лиц, проводивших испытание;
- Фамилий и инициалов лиц, подтвердивших проведение испытаний и результаты вычисле- ний, если требуется;
- Четкого заключения о выдаче разрешения или отклонении продукции (или другого решения о статусе продукции), даты и подписи ответственного лица;
- Перечня используемого оборудования.
-
- Все операции по внутрипроизводственному контролю, в т. ч. операции, выполняемые ли- цами, непосредственно работающими в производственных зонах, должны проводиться в соответст- вии с методиками, утвержденными отделом контроля качества, а их результаты должны быть оформ- лены документально.
- Особое внимание следует уделять качеству лабораторных реактивов, титрованных рас- творов, мерной лабораторной посуды, стандартных образцов и питательных сред. Их приготовление и подготовка должны соответствовать требованиям инструкций, утвержденных в установленном порядке.
*За исключением методов, установленных нормативными документами (прим. разработчика стандарта)
Степень контроля должна соответствовать их назначению и имеющимся данным о стабильности.
-
- Стандартные образцы должны соответствовать своему назначению. Подтверждение этому должно быть дано ясно и оформлено документально. Предпочтительно использовать в качестве пер- вичного стандартного образа фармакопейный стандартный образец из официально признанного ис- точника, если иное не является полностью обоснованным (допускается использование вторичных стандартов, если показана и задокументирована их прослеживаемость до первичного стандарта).
- Лабораторные реактивы, растворы, стандартные образцы и питательные среды, должны иметь маркировку с указанием даты приготовления и с подписями исполнителей. На этикетке должен быть указан срок годности реактивов, питательных сред и специфические условия их хранения. Для титрованных растворов следует указывать дату последнего установления титра и соответствующий поправочный коэффициент.
- При необходимости на таре следует указывать дату получения каждого вещества, исполь- зуемого для проведения испытаний (например, реактивов и стандартных образцов) с соответствую- щими инструкциями по их использованию и хранению. В некоторых случаях после получения или пе- ред использованием реактива может возникнуть необходимость проведения его испытания на под- линность и/или другого испытания.
- Питательные среды следует готовить в соответствии требованиями производителя, если иное не является научно обоснованным.
- Использованные микробиологические среды и споры следует инактивировать в соответст- вии с инструкцией и утилизировать с соблюдением мер предосторожности от перекрестных загрязне- ний и образования остатков. Сок хранения питательных сред до использования должен быть обосно- ван и документирован.
- Животных, используемых для контроля компонентов, материалов или продукции, следует, при необходимости, помещать в карантин перед началом работы с ними. Уход за животными и кон- троль за ними должны быть организованы так, чтобы обеспечить их пригодность к использованию по назначению. Животные должны быть маркированы, а истории работы с ними должны быть оформле- ны документально.
Программа последующих испытаний стабильности
-
- После реализации лекарственного средства следует систематически проводить испытания его стабильности по программе, позволяющей обнаруживать любые изменения, имеющие отношение к стабильности (например, изменение уровня примесей или профиля растворения) в соответствии с рецептурой реализованного продукта.
- Целью программы последующих испытаний стабильности является контроль самого про- дукта в течение его срока годности и установление того, что продукт соответствует (может быть при- знан соответствующим) спецификации при условиях хранения, указанных в маркировке.
- Вышеуказанные требования относятся, главным образом, к готовым лекарственным сред- ствам в упаковке, предназначенной для продажи, но следует также обратить внимание на включение в методику нерасфасованной продукции. Например, если нерасфасованную продукцию хранят дли- тельное время до упаковки и/или передачи с производственного участка на участок упаковки, то сле- дует изучить и оценить влияние этого фактора на стабильность готовой продукции при соответст- вующих параметрах окружающей среды. Более того, следует обратить внимание на промежуточную продукцию, которая хранится и используется в течение длительного периода. Исследование ста- бильности переработанных продуктов проводят при разработке лекарственного средства, последую- щего контроля стабильности такого продукта не требуется. Однако, при необходимости, может про- водиться контроль стабильности переработанного продукта.
- Программа последующих испытаний стабильности должна быть оформлена в докумен- тальном виде согласно общим правилам раздела 4, части 1, а результаты работы оформлены в виде отчета. Оборудование, используемое для последующих испытаний стабильности (в т. ч. камеры для контроля стабильности), должно быть аттестовано и обслуживаться в соответствии с общими прави- лами раздела 3, части I и приложения 15.
- Отчет о работе по программе последующих испытаний стабильности должен включать в себя весь период до истечения срока годности продукции и содержать следующие данные (но не ог- раничиваться ими):
i) номера серий для различных показателей эффективности и размеров серий, если требуется;
- данные о физических, химических, микробиологических и биологических методах испытаний;
- критерии приемлемости;
- ссылки на методы испытаний;
- описание упаковки и системы укупоривания;
- периодичность испытаний (сроки);
- описание условий хранения (следует использовать стандартные условия ICH* при длитель- ных испытаниях, соответствующих данным маркировки);
- другие показатели, относящиеся к данному лекарственному средству.
-
- Отчет о работе по программе последующих испытаний стабильности может отличаться от отчета о первоначальном длительном испытании стабильности, прилагаемому к регистрационному досье. Эти отличия должны быть обоснованы и указаны в отчете (например, периодичность контроля или проведение его в соответствии с рекомендациями ICH).
- Число серий и периодичность испытаний должны обеспечивать достаточный объем дан- ных для проведения анализа тенденций изменения. Если не указано иное, то в программу испытаний стабильности следует включить не менее одной серии произведенной продукции за год для каждого значения эффективности и вида первичной упаковки (если они различаются). Исключением являются случаи, когда в течение года данный продукт не выпускался. Для продуктов, последующие испытания стабильности которых требуют использования животных и не существует альтернативного аттесто- ванного метода, периодичность контроля может быть установлена с использованием анализа рисков. При наличии в отчете обоснования может использоваться принцип исключения из рассмотрения и построения матриц.
- В некоторых случаях в последующие испытания стабильности следует включать дополни- тельные серии. Например, последующие испытания стабильности следует проводить после любых существенных изменений или существенных отклонений в технологическом процессе или упаковке. Это же относится и к любым операциям по повторной обработке, переработке или регенерации.
- Результаты последующих испытаний стабильности должны быть доступны руководящим работникам, особенно уполномоченному лицу (лицам). Если последующие испытания стабильности проводят не на месте производства нерасфасованной или готовой продукции, то между участвующи- ми сторонами должно быть оформлено письменное соглашение. Результаты последующих испыта- ний стабильности должны находиться на месте производства для представления надзорным органам.
- Следует анализировать случаи выхода за пределы спецификации и существенные не- обычные тенденции. Любой подтвержденный выход за пределы спецификации или существенные негативные тенденции должны доводиться до сведения надзорных органов. Возможные решения в отношении выпущенных на рынок серий продукции следует принимать в соответствии с разделом 8, часть I по согласованию с надзорными органами.
- Следует документально оформлять и дополнять заключения по всем полученным данным, включая промежуточные выводы. Указанное заключение следует периодически рассматривать.
Порядок передачи методов испытаний
-
- До передачи метода испытаний передающая стороны должна проверить соответствие ме- тода испытаний методу, указанному в свидетельстве о регистрации или соответствующему техниче- скому досье. Следует периодически рассматривать данные об аттестации метода испытаний, чтобы убедиться в их соответствии действующим требованиям руководств ICH/VICH. При наличии пробелов следует провести дополнительный анализ с его документальным оформлением, чтобы установить необходимость в дополнительной аттестации до начала передачи методики.
- Передача аналитического метода от одной лаборатории (передающей) другой (прини- мающей) должна быть оформлена протоколом.
- Протокол передачи должен включать следующие пункты (но может не ограничиваться ими):
- Наименование (обозначение) испытания и передаваемого метода испытаний;
- Дополнительные требования к обучению;
- Стандарты и испытуемые образцы;
- Специальные требования к транспортированию и хранению испытуемых материалов;
- Критерии приемлемости, которые должны основываться на последних результатах аттеста- ции метода с учетом требований ICH/VICH.
-
- Следует рассмотреть отклонения от протокола передачи до завершения передачи методи- ки. Отчет о передаче методики должен содержать сравнение результатов процесса и указывать, в каких вопросах требуется провести дополнительную аттестацию метода, если требуется.
- При передаче метода испытаний следует учесть специальные требования других норма- тивных документов, если необходимо (например, к спектроскопии в ближней инфракрасной области).
* ICH – International Conference for Harmonization (Международная конференция по гармонизации). Здесь и далее по тексту (прим. разработчика стандарта).
7. Работа по контрактам на производство продукции и проведение анализов
Основные принципы
Во избежание разночтений, способных привести к ухудшению качества продукции или выпол- нения работ, следует тщательно составлять, согласовывать и контролировать выполнение контрак- тов на производство продукции и проведение анализов. Контракт между Заказчиком и Исполнителем должен быть оформлен в письменном виде с указанием четко определенных обязанностей каждой из сторон. В системе обеспечения качества Заказчика должен быть установлен порядок действий и от- ветственность уполномоченного лица за выдачу разрешения на реализацию каждой серии продукции.
П р и м е ч а н и е – Настоящий раздел рассматривает только ответственность производителей за выпол- нение требований настоящего стандарта, регистрационного досье и действующего законодательства.
Общие положения
-
- Контракт должен быть оформлен в письменном виде и должен включать в себя перечень производственных операций и/или анализов, выполняемых на основании положений контракта, и проводимых технических мероприятий.
- Выполнение контракта на производство и/или проведение анализов, в т. ч. с учетом пред- ложенных изменений технического или другого характера, должно соответствовать требованиям нормативных документов на производство и требованиям регистрационного досье.
- Если держатель свидетельства о регистрации и производитель не являются одним и тем же лицом, то следует предусмотреть меры согласно принципам данного раздела.
Заказчик
-
- Фармацевтическая система качества Заказчика должна предусматривать контроль и про- верку любой деятельности, переданной для выполнения другому лицу. Заказчик несет ответствен- ность за оценку компетентности данного лица и его способность выполнить контроль работ по кон- тракту, регламентируя его необходимыми мерами. К этим мерам относятся принципы анализа рисков и приведенные ниже требования:
- Заказчик несет ответственность за оценку До заключения контракта Заказчик правомочно- сти, способности и компетентности Исполнителя выполнить работы по контракту. Заказчик также не- сет ответственность, посредством контракта за выполнение требований GMP согласно настоящему стандарту.
- Заказчик предоставляет Исполнителю информацию, необходимую для выполнения преду- смотренных в контракте работ в соответствии с регистрационным досье и требований законодатель- ства. Заказчик должен убедиться в том, что Исполнитель обладает полной информацией о факторах, связанных с продукцией или работой, выполняемой по контракту, и представляющих опасность для его помещений, оборудования, персонала, других материалов или другой продукции.
- Заказчик должен контролировать и рассматривать работу Исполнителя и его деятельность по установлению необходимости изменений и их реализации.
- Заказчик несет ответственность за рассмотрение и оценку протоколов и результатов рабо- ты, относящихся к контракту. Он также должен убедиться сам или на основе подтверждения уполно- моченного лица Исполнителя о том, что вся продукция и материалы, поставленные ему Исполните- лем, произведены в соответствии правилами GMP и регистрационным досье.
Исполнитель
-
- Исполнитель должен иметь соответствующие помещения, оборудование, знания, опыт и компетентный персонал для надлежащего выполнения контракта.
- Исполнитель должен гарантировать пригодность получаемых им материалов, продукции и информации для использования по назначению.
- Исполнитель не должен привлекать третью сторону для выполнения работ, порученных ему по контракту, без предварительного рассмотрения и согласования с Заказчиком. При заключении соглашения между Исполнителем и третьей стороной должна быть обеспечена гарантия того, что информация, включая касающуюся оценки способности третьей стороны выполнить взятые не себя обязательства будут одинаково доступны первоначальному Заказчику и Исполнителю по контракту.
- Исполнитель не должен предпринимать действия, на которые он не уполномочен, за рам- ками контракта, способные оказать отрицательное влияние на качество работ для Заказчика.
- Исполнитель должен понимать, что работы по контракту, включая проведение анализов, могут явиться предметом проверки надзорными органами.
Контракт
-
- Контракт заключается между Заказчиком и Исполнителем. В контракте должны быть ука- заны ответственность сторон за производство и контроль продукции и порядок их общения. Техниче- ские аспекты контракта должны быть составлены компетентными лицами, обладающими соответст- вующими знаниями в области действия контракта и требований настоящего стандарта. Все действия по выполнению контракта должны соответствовать законодательству и требованиям, установленным при государственной регистрации лекарственного средства, и быть согласованы сторонами.
- В контракте должны быть четко указано, кто выполняет каждый пункт контракта, например, обучение, передачу технологии, цепь поставки, заключение субконтрактов, качество и приобретение материалов, испытания и выпуск материалов, производство и контроль качества (включая внутри- производственный контроль, отбор проб и проведение анализов).
- Вся документация, относящаяся к выполнению работ по контракту, например, на произ- водство, проведение анализов и реализацию, стандартные образцы, должны храниться или быть доступны Заказчику. Любые документы, относящиеся к контролю качества продукции должны, в слу- чае получения рекламаций или данных о возможном дефекте или для расследования в случае пред- полагаемой фальсификации продукции должны быть доступны Заказчику. Это должно быть отражено в инструкциях Заказчика.
- В контракте должно быть предусмотрено право Заказчика на аудит выполнения работ по контракту Исполнителем или субъектов, выполняющих работы по субконтракту и ранее взаимно со- гласованных.
8. Рекламации и отзыв продукции
Общие положения
С целью защиты здоровья человека и животных должна быть организована система с необхо- димой документацией для регистрации, оценки, расследований (рассмотрения) рекламаций, включая возможные дефекты, и, если требуется, быстрый и эффективный отзыв продукции для человека или животных и лекарственных средств для исследований из сети реализации. Следует применять прин- ципы анализа рисков к оценке и расследованиям дефектов и принятию решений по отзыву продукции, корректирующим и предупреждающим действиям и другим снижающим риск мерам. Руководство по применению этих принципов дано в разделе 1.
Надзорные органы должны быть своевременно извещены о подтвержденном дефекте (в произ- водстве, при изменениях свойств продукции, обнаружении фальсификации, несоответствия регист- рационному досье или спецификации или другой серьезной проблемы с качеством) лекарственного средства или лекарственного средства для исследований, которые могут привести к отзыву продук- ции или необычному ограничению на реализацию. Если обнаружено несоответствие находящейся на рынке продукции регистрационному досье, то нет требования извещать надзорный орган, если сте- пень несоответствия находится в пределах ограничений, установленных приложением 16 для дейст- вий при незапланированных отклонениях.
При работе по контракту в контракте должны быть указаны роль и ответственность производи- теля, держателя регистрационного досье и/или спонсора или третьей стороны в отношении оценки, принятия решения, распространения информации и применения действий по снижению риска в от- ношении дефектной продукции. Руководство по работе по контрактам дано в разделе 7. В таких кон- трактах должен быть предусмотрен порядок оповещения ответственных лиц каждой стороны для принятия мер в отношении дефекта и отзыва продукции.
Персонал и организация
-
- На предприятии должен быть персонал, имеющий достаточную квалификацию и опыт, кото- рый отвечает за работу с рекламациями и расследование дефектов и принимающий решения о свя- занных с ними мерах, включая отзыв продукции. Эти лица должны быть независимы от подразделе- ний реализации и маркетинга. Если в число этих сотрудников не входит уполномоченное лицо, отве- чающее за выпуск рассматриваемой серии (серий) продукции, то оно должно быть официально и без задержек извещено обо всех расследованиях и отзывах продукции.
- Предприятие должно располагать квалифицированным персоналом и ресурсами для обра- щения, оценки, расследований (рассмотрения) рекламаций и дефектов и принятию мер по снижению риска. Следует располагать квалифицированным персоналом и ресурсами для взаимодействия с компетентными органами.
- Следует привлекать специалистов разных направлений, включая подготовленный персонал по обеспечению качества.
- Если рассмотрение рекламаций и дефектов проводится на предприятии централизованно,
то следует документально определить роли других сторон. Центральный персонал не должен допус- кать задержек в данной работе.
Инструкции по работе и расследованию рекламаций, включая возможные де- фекты качества
-
- Должны быть письменные инструкции по действиям в случае поступления рекламации. Все рекламации должны документироваться и оцениваться на предмет наличия возможного дефекта или другого фактора.
- Следует обратить особое внимание на то, не является ли рекламация или возможный де- фект признаком фальсификации.
- Поскольку не все рекламации, получаемые предприятием, могут действительно быть связа- ны с дефектом, следует вести отдельную документацию на такие рекламации и ставить в известность о них лиц, отвечающих за расследования с целью установления возможных отрицательных событий.
- Должны быть по анализу качества серии лекарственного средства для расследования воз- можного отрицательного события, при поступлении соответствующего запроса.
- Инструкция по расследованию дефекта должны включать:
- Описание дефекта;
- Определение масштаба дефекта. Проверка или испытания контрольных и/или архивных об- разцов и, в некоторых случаях, анализ протоколов серии, документации о выпуске и реализации се- рии (особенно для чувствительной к температуре продукции);
- Указание на необходимость запроса образца или возврата продукции от подавшего рекла- мацию лица и, если образец представлен, его оценку;
- Оценку риска от дефекта исходя из его тяжести и масштаба;
- Принятие решения с учетом необходимости снижения риска в сети реализации, например, об отзыве серии или другие действия;
- Оценка влияния отзыва продукции на ее доступность на рынке для людей или животных и необходимости извещения надзорных органов о таком влиянии;
- Внешние и внутренние взаимодействия, которые следует осуществить в отношении дефек- та и его расследования;
- Указание на вероятную причину дефекта;
- Необходимость в определении и принятии корректирующих и предупреждающих действий (САРА) и оценки их эффективности.
Расследование и принятие решения
-
- Следует документально оформить информацию о возможном дефекте, включая все пер- вичные данные. Следует оценить достоверность информации и масштаб дефектов в соответствии принципами анализа рисков в качестве поддержки решений в отношении расследований и действий. Результаты следует оформить документально.
- Если в серии обнаружен дефект или есть подозрение о нем, то следует рассмотреть во- прос о проверке других серий, в некоторых случаях, другой продукции на предмет наличия в них де- фектов. В отношении серий, которые могут сдержать части дефектных серий, должно быть проведено расследование.
- Расследование дефектов должно включать анализ прежних отчетов о дефектах или другой относящейся к предмету информации с целью обнаружения специфических или повторяющихся про- блем, требующих внимания или возможных действий со стороны надзорных органов.
- Решения, принимаемые в ходе расследования дефекта и после него должны отражать уровень риска, вызываемого дефектом, а также серьезность любого несоответствия требованиям свидетельства о регистрации, спецификации на продукцию или правилам GMP. Такие решения долж- ны приниматься своевременно, чтобы гарантировать безопасность пациента или животного с учетом риска, представляемого такими факторами.
- Полная информация о природе и масштабах дефекта не всегда может быть известна на ранних стадиях расследования. В связи с этим процесс принятия решения должен включать действия по снижению риска на соответствующем этапе расследования. Все решения, принимаемые в связи с дефектом меры должны оформляться документально.
- Производитель должен своевременно извещать надзорный орган, выдавший свидетельст- во о регистрации, спонсора и все имеющие отношения к вопросу компетентные органы в случаях, ко- гда дефект может привести отзыву продукции или ненормальному ограничению в ее применении.
Поиск причин дефекта и предупреждающие и корректирующие действия
-
- При расследовании дефекта следует найти его причину. Если истинную причину найти не удается, то следует установить наиболее вероятную причину и провести их анализ.
- Если причиной дефекта является установлена ошибка человека или есть подозрение на нее, то это должно быть формально обосновано и следует выполнить анализ процесса, документа- ции, системных ошибок или проблем на предмет наличия упущений.
- Следует определить и предпринять корректирующие и предупреждающие действия (СА- РА) в отношении дефекта. Следует контролировать и оценивать эффективность этих действий.
- Следует регулярно рассматривать документацию о дефектах на предмет и выполнять ана- лиз тенденций для обнаружения специфических или повторяющихся проблем, требующих внимания.
Отзывы продукции и другие действия по снижению риска
-
- На предприятии должны письменные инструкции по отзывам продукции или принятию дру- гих мер по снижению риска, которые должны регулярно оцениваться и пересматриваться, при необ- ходимости.
- После отгрузки продукции любое изъятие его из сети реализации из-за дефекта должно рассматриваться как отзыв (это не распространяется на изъятие или возврат образов продукции из сети реализации для проведения исследований или подготовке отчета в связи с дефектом.
- Отзыв продукции должен осуществляться оперативно и в любое время. В некоторых слу- чаях отзыв может потребоваться для защит здоровья человека или животных до выяснения причины дефекта и получения полных данных о нем.
- Документация по отгрузке серии продукции должна быть доступной для лица (лиц), ответ- ственного(ых) за отзыв продукции, и содержать достаточную информацию об оптовых покупателях и прямых заказчиках (адреса, номера телефонов/факсов в рабочее и в нерабочее время, номера серий и объемы поставок), экспортных поставках и поставках образцов лекарственных средств.
- В случае лекарственных средств для исследований должны быть идентифицированы все места проведения испытаний с указанием стран. Если для лекарственного средства для исследова- ний выдано разрешение на применение, то его производитель совместно со спонсором должен по- ставить в известность держателя этого разрешения о любых дефектах, относящихся этому продукту. Спонсор должен немедленно принять меры к снятию маскировки маскированной продукции и ее от- зыву, при необходимости.
- Следует, если требуется, провести консультации с компетентными органами о порядке от- зыва, учитывая возможный риск для здоровья человека или животных и любые последствия, вызван- ные отзывом. Следует также информировать компетентные органы в случаях, когда не предусматри- вался отзыв дефектной серии ввиду истечения срока годности (например, для продукции с коротким сроком годности).
- Компетентные органы стран, куда могла быть направлена продукция, должны быть зара- нее информированы о намерении отзывать продукцию. В каждом серьезном случае (например, когда возможный дефект может вызвать последствия для здоровья человека или животного), могут потре- боваться быстрые действия по снижению риска (например, отзыв продукции) до извещения компе- тентного органа. По возможности следует принять меры по их выполнению с указанным компетент- ным органом.
- Следует учесть последствия, к которым может привести предлагаемый отзыв продукции с рынка и, если они возможны, то разработать специфические меры по снижению риска на рынке и рассмотрены с компетентным органом. Учитывая во внимание использование продукта в терапевти- ческих целях, следует оценить риск прекращения его выпуска, если для продукта нет разрешенной альтернативы, до принятия решения о таких мерах по снижению риска как отзыв продукции. Любые решения не принимать меры по снижению риска, которое в другом случае потребовались бы, следует заранее согласовать с компетентным органом.
- На отозванной продукции должна быть соответствующая маркировка. Отозванную продук- цию следует хранить в надежно изолированных зонах до принятия решения в отношении ее. Место нахождения всех отозванных серий должно быть оформлено документально. Обоснование решения о переработке отозванной продукции должно быть документировано и рассмотрено соответствующим компетентным органом. Следует также учесть оставшийся срок хранения переработанной серии, ко- торую предполагается реализовать.
- Последовательность действий при отзыве продукции должна быть оформлена докумен- тально. Окончательный отчет должен содержать баланс между количеством поставленной и отозван- ной продукции.
- Эффективность мероприятий по отзыву продукции следует регулярно анализировать и подтверждать ее неизменность. Такие оценки следует проводить как в рабочее, так и нерабочее вре- мя, при этом следует учесть необходимость в проведении тренировочного отзыва.
- В дополнение к отзывам продукции существуют другие меры по снижению риска, которые могут быть применены к рискам, вызываемым дефектами. Такие меры могут включать рассылку пре- дупреждений специалистам, занятым в сфере здравоохранения, в отношении использования серий с возможными дефектами. Это следует рассматривать в индивидуальном порядке и обсуждать с ком-
петентными органами.
9. Самоинспекции
Общие положения
Самоинспекция (внутренний аудит) должна проводиться с целью проверки выполнения предпри- ятием требований настоящего стандарта и принятия необходимых мер по устранению недостатков.
-
- С целью обеспечения качества продукции вопросы, связанные с работой персонала, поме- щениями, оборудованием, документацией, производством, контролем качества и реализацией лекар- ственных средств, мероприятиями по работе с рекламациями и отзыву продукции, а также проведе- нию самоинспекций, должны регулярно рассматриваться в соответствии с утвержденной программой и принципами обеспечения качества.
- Самоинспекция должна проводиться независимо и тщательно специально назначенным со- трудником(ами), состоящим(ими) в штате предприятия. Может быть полезным проведение независи- мого аудита экспертами сторонних организаций.
- Результаты самоинспекций должны быть оформлены документально. Протоколы (отчеты), составленные по результатам самоинспекции, должны включать в себя всю полученную информацию и необходимые корректирующие действия (при необходимости). Действия, предпринимаемые по ре- зультатам проведенной самоинспекции, следует оформлять документально.
В настоящем стандарте используются следующие термины с соответствующими определениями:
- 1 аттестация; испытания (qualification, validation): Доказательство того, что методика, процесс, оборудование, материал, операция или система соответствуют заданным требованиям и их использование действительно дает ожидаемые результаты.
- 2 баллон (cylinder): Сосуд для хранения газа при высоком давлении.
- 3 банки клеток (cell bank):
- система банков клеток (cell bank system):Система, при которой последовательные серии продукции производят из клеточных культур, принадлежащих главному банку клеток, который полностью охарактеризован на подлинность и отсутствие загрязнений. Некоторое количество емкостей главного банка клеток используется для формирования рабочего банка клеток. Система банков клеток должна быть аттестована (испытана) на определенное количество пересевов или количество удвоений популяции, до достижения которых они могут использоваться в текущем производстве.
- главный банк клеток (master cell bank): Культура клеток, полностью охарактеризованная на подлинность и отсутствие загрязнений, распределенная по емкостям в процессе одной операции таким образом, чтобы обеспечивались ее однородность и стабильность. Как правило, главный банк клеток хранится при температуре минус 70 °С или ниже.
- рабочий банк клеток (working cell bank): Культура клеток, отобранная из главного банка клеток для приготовления производственных культур клеток. Рабочие банки клеток хранят при температуре минус 70 °С или ниже.
- 4 биологические агенты (biological agents): Микроорганизмы, в т. ч. полученные методами генной инженерии, культуры клеток и эндопаразиты, патогенные или непатогенные.
- 5 биореактор (biogenerator): Изолированная система (например, ферментатор), в которую вместе с другими материалами вводят биологические агенты и в результате протекающей реакции происходит их размножение или образование других веществ. Биореакторы, как правило, оборудуются устройствами для регулирования, контроля, добавления и извлечения материалов.
- 6 внутрипроизводственный контроль (in-process control): Контроль, выполняемый в ходе технологического процесса с целью проверки соответствия продукции заданным требованиям, по результатам которого может выполняться корректировка параметров технологического процесса. Контроль состояния окружающей среды или оборудования рассматривается как элемент внутрипроизводственного контроля.
- 7 возврат (return): Возврат лекарственного средства его производителю или поставщику.
- 8 воздушный шлюз (air-lock): Ограниченное пространство с двумя или несколькими дверями между двумя или несколькими помещениями (например, различных классов чистоты), предназначенное для разделения воздушных сред помещений при входе в них. Воздушный шлюз служит для перехода персонала или перемещения материалов.
- 9 готовая продукция (готовый продукт) (finished product): Лекарственное средство, прошедшее все этапы технологического процесса, в т. ч. окончательную упаковку.
- 10 изолированная зона (contained area): Зона, оборудованная соответствующими фильтрами и устройствами подготовки воздуха для предотвращения загрязнения внешней окружающей среды биологическими агентами, присутствующими в этой зоне.
- 11 изоляция (containment): Меры по ограничению распространения биологического агента или другого вещества за пределы определенного пространства.
- первичная изоляция: Изоляция, препятствующая прониканию биологического агента в окружающую среду, непосредственно прилегающую к рабочей зоне. Предусматривает использование закрытых контейнеров или безопасных биологических шкафов и методов безопасного выполнения технологических операций.
- вторичная изоляция: Изоляция, препятствующая прониканию биологических агентов во внешнюю окружающую среду или в другие рабочие зоны. Предусматривает использование помещений со специальными системами подготовки воздуха, воздушных шлюзов и/или стерилизаторов для перемещения материалов и обеспечивает безопасное выполнение технологических операций. Во многих случаях используется для повышения эффективности первичной изоляции.
- 12 инструкция; методика; процедура (procedures): Документ, содержащий указания по выполнению отдельных видов операций (например, по очистке, переодеванию, контролю окружающей среды, отбору проб, проведению испытаний, эксплуатации оборудования).
- 13 инфицированный (infected): Зараженный чужеродными биологическими агентами и способный к распространению инфекции.
- 14 испытания (validation): См. аттестация.
- 15 исходные материалы (starting material): Любое вещество, используемое для производства лекарственных средств, кроме упаковочных материалов.
- 16 калибровка; поверка (calibration): Операции, устанавливающие при определенных условиях зависимость между значениями, регистрируемыми контрольно-измерительными приборами (системами) и соответствующими стандартными величинами (эталонами).
- 17 карантин (quarantine): Статус исходных или упаковочных материалов, промежуточной, нерасфасованной или готовой продукции, изолированных физически или иным образом до вынесения решения об их выпуске или отклонении.
- 18 коллектор (manifold): Устройство или оборудование, позволяющее одновременно наполнять газом несколько баллонов (контейнеров) из одного источника.
- 19 контролируемая зона (controlled area): Зона, построенная и эксплуатируемая таким образом, чтобы предотвратить внесение возможного загрязнения и случайное распространение живых организмов (например, может иметь систему воздухоподготовки, соответствующую зоне D). Степень контроля зависит от свойств организмов, используемых в технологическом процессе. Как минимум, такая зона должна иметь отрицательное давление по отношению к смежным помещениям и возможность эффективно удалять незначительные количества аэрозольных загрязнений.
- 20 контроль качества (quality control): См. пункт 1.4 раздела 1.
- 21 криогенный сосуд (cryogenic vessel): Сосуд для хранения сжиженных газов при сверхнизких температурах.
- 22 культура клеток (cell culture): Клеточная масса, полученная в результате выращивания in vitro клеток, изолированных от многоклеточных организмов.
- 23 лекарственное растение (medicinal plant): Растение или часть его, используемое для медицинских целей.
- 24 лекарственное средство из растительного сырья (herbal medicinal product): Лекарственное средство, содержащее в качестве активных ингредиентов только вещества растительного происхождения и/или препараты на их основе.
- 25 нерасфасованный готовый продукт балк-продукт (bulk product): Продукт, прошедший все производственные стадии, за исключением окончательной упаковки.
- 26 номер серии; номер партии (batch number or lot number): Определенное сочетание цифр и/или букв, обозначающее серию продукции.
- 27 перекрестное загрязнение (cross contamination): Загрязнение материалов или продукции другими материалами или продукцией.
- 28 переработка (reprocessing): Повторная обработка серии или части серии продукции, не соответствующей заданным требованиям, начиная с определенной стадии производства, для получения продукции требуемого качества после проведения одной или нескольких дополнительных операций.
- 29 повторное использование (recovery): Включение произведенной ранее серии продукции требуемого качества (или ее части) в другую серию продукции на определенной стадии производства.
- 30 посевной материал (seed lot):
- система посевных материалов (seed lot system): Система, при которой последовательные серии продукции получают из главного посевного материала при определенном количестве пересевов (пассажей). В текущем производстве рабочий посевной материал готовится из главного посевного материала. Готовый продукт производится из рабочего посевного материала, при этом число пересевов из главного посевного материала не должно превышать значения, установленного при клинических испытаниях вакцин, исходя из требований безопасности и эффективности. Происхождение главного посевного материала и история пересевов из него должны оформляться документально.
- главный посевной материал (master seed lot): Культура микроорганизмов, распределенная из одного нерасфасованного продукта по емкостям в процессе одной операции таким образом, чтобы обеспечивались однородность, стабильность и не допускалось загрязнение. Главный посевной материал в жидком виде хранят при температуре минус 70 °С; в лиофилизированном виде – при температуре, обеспечивающей его стабильность.
- рабочий посевной материал (working seed lot): Культура микроорганизмов, полученная из главного посевного материала и предназначенная для использования в производстве. Рабочий посевной материал распределяется по емкостям и хранится аналогично хранению главного посевного материала.
- 31 производитель (manufacturer): Держатель лицензии на производство*.
- 32 производство (manufacture): Все операции и виды контроля, связанные с получением, приемкой и обработкой исходных материалов, упаковкой, выпуском в реализацию, хранением и отгрузкой лекарственных средств.
- 33 промежуточный продукт (intermediate product): Частично обработанный материал, который должен пройти дальнейшие стадии производства, прежде чем он станет нерасфасованным готовым продуктом.
- 34 протокол на серию (record): См. раздел 4.
* Предприятие, осуществляющее хотя бы один этап производства, рассматривается как производитель лекарственных средств (прим. разработчика стандарта). - 35 радиофармацевтический препарат (radiopharmaceutical): Любое лекарственное средство, содержащее в готовом виде один или более радионуклидов (радиоактивных изотопов), используемых для медицинских целей.
- 36 растительное сырье (crude plant; vegetable drug): Сырые или высушенные лекарственные растения или их части.
- 37 серия; партия (batch or lot): Определенное количество однородных исходных и упаковочных материалов или однородной продукции, обработанной в ходе одной или нескольких последовательных технологических стадий.Примечание – При необходимости на определенных стадиях производства серия может быть разделена на подсерии, объединяемые впоследствии в однородную серию продукции. При непрерывном производстве понятие серии должно относиться к определенной части продукции, характеризуемой однородностью. С точки зрения контроля готовой продукции серия продукции включает в себя совокупность единиц дозированной формы лекарственных средств (лекарственной формы), изготовленных из одного объема исходного материала и прошедших единую последовательность производственных операций или единый цикл стерилизации; при непрерывном производстве – все единицы, произведенные в заданный интервал времени.
- 38 сжиженные газы (liquifiable gases): Газы, которые при стандартных температуре и давлении наполнения находятся в баллоне в сжиженном виде.
- 39 система (system): Совокупность взаимосвязанных действий и технических средств, образующих единое целое.
- 40 система с компьютерным управлением и контролем (computerized system): Система, включающая в себя ввод данных, их электронную обработку и вывод информации, используемой для регистрации или автоматического контроля.
- 41 сопоставление; выход продукции (reconciliation): Сравнение ожидаемого и фактического объемов произведенной или использованной продукции с учетом стандартных отклонений.
- 42 спецификация (specification): См. раздел 4.
- 43 стерильность (sterility): Отсутствие живых микроорганизмов. Требования к проведению контроля стерильности приведены в соответствующей нормативной документации.
- 44 технологический процесс (production): Операции, включающие в себя приемку и обработку исходных материалов, упаковку и получение готового продукта.
- 45 упаковка (packaging): Все операции, в т. ч. наполнение и маркировка, проводимые с нерасфасованным продуктом для получения готового продукта. Примечание – Стерильное наполнение, как правило, не следует рассматривать как часть процесса упаковки. В этом случае нерасфасованным продуктом считаются наполненные первичные контейнеры без окончательной упаковки.
- 46 упаковочный материал (packaging material): Любой материал, применяемый для упаковывания лекарственных средств, за исключением внешней упаковки, используемой для транспортирования. Упаковочные материалы делятся на первичные или вторичные в зависимости от наличия прямого контакта с продуктом.
- 47 чистая зона (clean area): Зона, построенная и эксплуатируемая таким образом, что в ней сведено к минимуму проникание, образование и накопление загрязнений в виде частиц и микроорганизмов. Примечание – Типы чистых зон определены в приложении 1.
- 48 чистая изолированная зона (clean/contained area): Зона, построенная и эксплуатируемая таким образом, что она одновременно является чистой и изолированной зоной.
- 49 экзотический организм (exotic organism): Биологический агент, вызывающий заболевание, отсутствующее в данной стране или географической зоне, либо являющийся объектом профилактических мер или программы по его устранению.
1 Основные принципы
- Лекарственные средства должны соответствовать утвержденным спецификациям и требованиям настоящего стандарта (GMP). Их выпуск в реализацию допускается, как правило, после завершения видов контроля активных субстанций и/или готовой продукции в соответствии с регистрационным досье или протоколом клинических исследований. В некоторых случаях вместо контроля конечного продукта допускается выпуск серии на основе данных о продукте, процессе и информации, полученной в ходе технологического процесса, при получении на это разрешения. Любые действия, требуемые для этой формы выпуска серии, должны быть включены в фармацевтическую систему качества.
2 Область применения
- Настоящее приложение устанавливает требования к выпуску в реальном времени и выпуску по параметрам, когда контроль критических параметров и свойств материалов служит альтернативой обычному контролю активной субстанции и/или готовой продукции, если разрешение на это получено. Оно предусматривает применение контроля в реальном времени к любой стадии технологического процесса и любой активной субстанции, готовой или промежуточной продукции.
3 Контроль в реальном времени
- Контроль выпуска в реальном времени предусматривает внутрипроизводственный контроль и проверки, которые заменяют контроль готовой продукции, как условие для выпуска продукции (если это разрешено). Для применения этого метода следует получить разрешение надзорного органа и информировать его об оценке процесса в ходе текущей работы. Степень взаимодействия с надзорным органом зависит от сложности контроля в реальном времени.
- Результаты контроля параметров и свойств материалов в реальном времени должны точно отражать свойства готовой продукции;
- Следует определить свойства материалов и методы контроля процесса, которые позволят заменить контроль готовой продукции с научным обоснованием на основе знаний о материалах, продукте и процессе;
- Четким обоснованием возможности контроля выпуска в реальном времени и решения о выпуске серии должны быть комбинированный контроль процесса (параметров процесса и свойств материалов) и другие данные, полученные в ходе производства.
- Контроль выпуска в реальном времени должен быть включен и подлежит контролю в рамках фармацевтической системы качества. Информация о нём должна включать, как минимум, следующее:
- анализ рисков, в том числе для всего процесса, в соответствии с разделом 1 Части I и разделом 2 Части II настоящего стандарта;
- порядок контроля изменений;
- стратегию контроля;
- специальную программу обучения персонала;
- порядок проведения аттестации (испытаний);
- систему корректирующих и предупреждающих действий (CAPA) при отклонениях;
- действия в случае отказа сенсоров или оборудования;
- периодическое рассмотрение эффективности контроля выпуска в реальном времени в плане обеспечения качества продукции.
- Контроль изменений является важной частью метода контроля выпуска в реальном времени в соответствии с принципами раздела 1 Части I, раздела 13 Части II и приложения 15 к настоящему стандарту. Любые изменения, которые могут оказать влияние на производство и контроль продукции или статус «аттестован» помещений, оборудования, систем, аналитических методов или процессов, следует оценивать в плане риска для качества продукции и влияния на воспроизводимость технологического процесса. Следует обосновывать любые изменения путем ясного применения анализа рисков с полным документальным оформлением. После внесения изменений следует их оценить и показать, что они не привели к непредусмотренному или отрицательному влиянию на качество продукции.
- Порядок контроля должен быть разработан не только для контроля процесса, но и для подтверждения постоянного качества выпускаемой продукции. В стратегию контроля должны быть включены внутрипроизводственный контроль, контроль свойств материалов и параметров процесса, которые подлежат текущему контролю и должны быть основаны на понимании производства и процесса. Стратегия контроля может претерпевать изменения в течение жизненного цикла продукции и требовать применения анализа рисков и получения новой продукции. В стратегию контроля также должен входить план отбора проб и критерии приемки и отклонения.
- Персонал должен пройти специальное обучение по контролю выпуска в реальном времени, его принципам и методам. Основной персонал должен обладать опытом, знать и понимать продукт и процесс. Для успешного контроля выпуска в реальном времени нужна совместная работа представителей разных специальностей, обладающих знаниями в специальных областях, включая инженерные системы, аналитические методы, хемометрическое моделирование и статистику.
- Важными частями контроля выпуска в реальном времени являются аттестация (испытания) и, особенно, современные аналитические методы. Особое внимание следует обращать на аттестацию (испытания), контроль и регулирование «на линии», аналитические методы контроля «на линии», когда пробоотборник установлен в технологическом оборудовании.
- Следует тщательно анализировать любые отклонения или сбои в процессе и принимать меры по корректировке отрицательных изменений.
- Важную роль играет постоянный сбор и анализ данных в течение жизненного цикла продукта, который входит в фармацевтическую систему качества. Современная техника позволяет обнаруживать тенденции изменения параметров, отрицательно влияющих на процесс. Производители должны оценивать данные с научной точки зрения, консультируясь, при необходимости, с надзорными органами, чтобы определить, как анализ тенденций позволяет установить возможность повышения качества и/или поддержания его на требуемом уровне.
- После утверждения порядка контроля выпуска в реальном времени его следует использовать при текущем выпуске продукции. Если результаты контроля отрицательные или наблюдается тенденция к нарушению требований, то данный подход не может быть заменен на контроль серии готовой продукции. Любая ошибка должна быть тщательно расследована, а принятие решения о выпуске серии быть основано на результатах данного расследования и соответствовать регистрационному досье и требованиям GMP. Тенденции следует отслеживать соответствующим образом.
- Свойства (например, однородность содержания) не контролируются непосредственно при утвержденном порядке контроля выпуска в реальном времени, и должны по-прежнему указываться в Паспорте анализа серии. Следует указать утвержденный метод контроля готовой продукции и заключение «Соответствует, если проверено» с примечанием внизу: «Проверено с применением утвержденного метода контроля выпуска в реальном времени».
4 Выпуск по параметрам и стерилизация
- Данный раздел устанавливает требования к выпуску по параметрам, который определяется как выпуск серии продукции, прошедшей финишную стерилизацию, на основе рассмотрения результатов контроля критических параметров процесса, а не по данным контроля стерильности готовой продукции.
- Возможности обнаружения загрязнений при контроле готовой продукции на стерильность ограничены ввиду того, что контролю подлежит лишь малая доля от всей серии и на питательной среде могут прорасти лишь некоторые, а не все микроорганизмы. В связи с этим контроль готовой продукции на стерильность может выявить лишь грубый недостаток в обеспечении стерильности (например, недостаток, который привел к загрязнению большого числа единиц продукции и/или загрязнению специфическими микроорганизмами, рост которых поддерживается заданной питательной средой). В противоположность этому данные, полученные путем внутрипроизводственного контроля (например, контроль бионагрузки перед стерилизацией или контроль окружающей среды) и путем контроля параметров процесса стерилизации, которые могут предоставить более точную информацию для обеспечения стерильности продукции.
- Выпуск по параметрам может применяться только для продукции, стерилизуемой в окончательной упаковке влажным теплом, сухим теплом или ионизирующим излучением (дозиметрический выпуск) в соответствии с установленными требованиями.
- Для применения этого метода производитель должен располагать предшествующими данными о соответствии требованиям GMP и четкой программой обеспечения стерильности для подтверждения того, что процесс постоянно находится под контролем и есть понимание процесса.
- Программа обеспечения стерильности должна быть оформлена документально и включать в себя, как минимум, контролируемые критические параметры процесса, описание процесса стерилизации и ее аттестацию, аттестацию целостности контейнера (первичного упаковочного материала), методов контроля биозагрязнений и окружающей среды, принципа разделения продукции, проектные решения по оборудованию, обслуживающих систем и помещений и программу аттестации, инструкции по техническому обслуживанию, порядок калибровки, инструкцию по внесению изменений, программу обучения персонала и данные по анализу рисков.
- Анализ рисков является важным требование для выпуска по параметрам и должен быть направлен на устранение факторов, которые повышают риск нестерильности любой единицы продукции в каждой серии. Если предполагается выпуск по параметрам новой продукции или для нового процесса, то оценка риска должна быть выполнена на стадии разработки процесса, включая оценку данных о существующей продукции, если это применимо. Если учитываются существующая продукция или процесс, то анализ рисков должен включать оценку предшествующих данных.
- Персонал, участвующих в выпуске по параметрам, должен иметь опыт работы в следующих областях: микробиология, обеспечение стерильности, технические системы, производство и стерилизация. Следует документально оформлять сведения о квалификации, опыте, компетентности и прохождении обучения всех лиц, участвующих в выпуске по параметрам.
- Любые предлагаемые изменения, которые могут оказать влияние на обеспечение стерильности, должны регистрироваться в системе внесения изменений и рассматриваться лицами, имеющими подготовку и опыт в области обеспечения стерильности.
- Для обеспечения выпуска по параметрам следует разработать программу контроля бионагрузки продукции и компонентов до стерилизации. Контроль бионагрузки следует выполнять для каждой серии. Точки отбора проб в наполненных единицах до стерилизации отражать условия наихудшего случая и быть представительными для всей серии. Следует идентифицировать любые организмы, обнаруженные при контроле бионагрузки, с целью подтверждения того, что они не являются спорообразующими и могут быть более устойчивыми к процессу стерилизации.
- Бионагрузка на продукции должна быть сведена к минимуму при проектировании производственной среды и процесса за счет:
- правильных проектных решений оборудования и помещений, которые должны обеспечить эффективную очистку и дезинфекцию;
- подробных и эффективных методов очистки и дезинфекции;
- использования фильтров, удерживающих микроорганизмы, где это возможно;
- инструкций и методик, направленных на обеспечение гигиены персонала и требуемой одежды;
- установления пределов на микробные загрязнения исходных материалов, промежуточной продукции и вспомогательных сред (например, газов).
Процесс стерилизации
- Аттестация (испытания) играют критическую роль в подтверждении того, что оборудование для стерилизации может постоянно обеспечивать параметры цикла стерилизации и устройства мониторинга обеспечивают контроль процесса стерилизации.
- Следует периодически проводить повторную аттестацию в соответствии с приложениями 1 и 15 к настоящему стандарту.
- Измерение критических параметров в ходе процесса стерилизации является критическим требованием для выпуска по параметрам. Следует задать требования к контрольно-измерительным приборам для процесса стерилизации. Калибровка приборов должна быть прослеживаема до соответствующих стандартов.
- Следует определить критические параметры процесса и периодически их оценивать. Следует определить пределы изменения параметров на основе требований к процессу, характеристик процесса, допустимых значений при калибровке и степени критичности параметра.
- При текущем контроле стерилизатора должно быть показано, что для каждого цикла выполняются условия, которые нужны для специфического процесса и которые были установлены при аттестации процесса. В течение цикла стерилизации следует контролировать критические параметры
- Документация (протокол) стерилизации должна включать все критические параметры процесса. Эту документацию следует проверять на соответствие спецификации по крайней мере по двум независимым каналам (двумя лицами или аттестованной компьютерной системой и одним лицом).
- После того, как надзорным органом выдано разрешение на выпуск по параметрам, решение о выпуске или отклонении серии следует принимать в соответствии с утвержденными спецификациями и на основе рассмотрения данных контроля критических параметров процесса. Результаты текущего контроля стерилизатора, изменений, отклонений, планового не непланового технического обслуживания следует оформлять документально, оценивать и утверждать до выпуска продукции в реализацию. Несоответствие спецификации при выпуске по параметрам не может быть компенсировано успешным результатом контроля стерильности готовой продукции.
5 Термины
Стратегия контроля (Control Strategy): Плановый комплекс мер по контролю, построенный исходя из понимания продукта и процесса в текущее время, обеспечивающий выполнение требований к процессу и качеству продукции. Эти меры могут включать контроль параметров и свойств, относящихся к субстанциям лекарственных средств, материалов и компонентов лекарственных средств, условий работы помещений и оборудования, внутрипроизводственный контроль, спецификации на готовую продукцию и связанные с ними методы и периодичность контроля. [ICH Q10] Критический параметр процесса (critical process parameter): Параметр процесса, изменение которого влияет на критическое свойство для качества и который в связи с этим следует контролировать или регулировать для обеспечения выпуска продукции заданного качества. [ICH Q8 (R2)] Критическое свойство для качества (critical quality attribute): Физическое, химическое, биологическое или микробиологическое свойство или характеристика, которая должна находиться в заданных пределах (диапазоне значений) или распределения с целью обеспечения требуемого качества продукции. [ICH Q8 (R2)] Выпуск по параметрам (parametric release): Одна из форм контроля выпуска в реальном времени. Выпуск по параметрам продукции, подлежащей финишной стерилизации, основан на рассмотрении данных о контроле процесса (например, температуры, давления времени финишной стерилизации), а не на контроле образцов для оценки критического свойства (Это совместно с выполнением специальных требований GMP к выпуску по параметрам обеспечивает требуемое качество продукции). [ICH Q8 Q&A] Контроль выпуска в реальном времени (real time release testing): Возможность оценивать и гарантировать качество промежуточной и/или готовой продукции на основе данных о процессе, что обычно включает в себя сочетание данных контроля свойств материала и процесса. [ICH Q8] Состояние контроля (State of Control): Условия, при которых комплекс мер по контролю последовательно обеспечивает выполнение процесса и требований к качеству. [ICH Q10]Приложение 14. Правила GMP EC 2019 (Производство лекарственных средств из крови или плазмы человека)
Термины и определения
Кровь (blood): Цельная кровь, заготовленная от одного донора и обработанная для переливания или дальнейшего производства. Компоненты крови (blood components): Используемые для лечебных целей составляющие крови (эритроциты, лейкоциты, плазма, тромбоциты), которые могут быть приготовлены центрифугированием, фильтрацией и замораживанием с использованием методов, установленных в банке крови. Организация по взятию и проверке крови (establishment): организация, которая отвечает за любой аспект взятия и проверки донорской крови или компонентов крови независимо от их дальнейшего предназначения, а также за их обработку, хранение и поставку в случае, когда они предназначены для трансфузии. Этот термин не распространяется на банки крови в больницах, но распространяется на организации, в которых проводят плазмаферез; Препараты крови (medicinal products derived from blood or plasma; blood product*): Лекарственные средствf, получаемst из крови или плазмы человека. Фракционирование, предприятие по фракционированию (fractionation, fractionation plant): технологический процесс (предприятие по фракционированию), во время которого выделяют компоненты плазмы с помощью различных физических и химических методов, например, осаждение, хроматография. Лекарственные средства, получаемые из донорской крови или плазмы (medicinal products derived from human blood or human plasma): лекарственные средства основе компонентов крови, произведенные промышленным способом на государственных или частных предприятиях. Плазма для фракционирования (plasma for fractionation): жидкая часть донорской крови, которая остается после отделения клеточных компонентов крови, отобранная в контейнер с антикоагулянтом, либо которая остается после сепарации с помощью непрерывной фильтрации или центрифугирования крови с антикоагулянтом во время процесса афереза. Плазма для фракционирования предназначена для производства лекарственных средств, получаемых из плазмы, например, альбумина, факторов свертываемости и иммуноглобулинов человека, которые описаны в Государственной фармакопее Российской Федерации. Основное досье на плазму (Plasma Master File): отдельный документ, который не входит в регистрационное досье на лекарственное средство. Этот документ содержит всю необходимую подробную информацию в отношении характеристик цельной донорской плазмы, используемой как исходный материал и/или сырье для производства промежуточных фракций, вспомогательных веществ и активных фармацевтических субстанций, которые являются частью плазмы, лекарственных средств или медицинских изделий. обработка (processing): любой из этапов получения компонентов крови, который осуществляется после взятия крови перед получением компонента крови, например, сепарация и замораживание компонентов крови. В настоящем приложении под обработкой дополнительно понимают выполняемые в организациях по взятию крови операции, которые являются специфическими для плазмы, используемой для фракционирования. Уполномоченное лицо (qualified person): см. приложение 16 к настоящему стандарту. Ответственное лицо (Responsible Person): специально назначенное лицо в организации по взятию и проверке крови, которое несет ответственность за:- обеспечение того, что кровь или ее компоненты были взяты и проверены в каждой единице независимо от их предназначения, а также за то, что (в случае предназначения для трансфузии) их обработка, хранение и отпуск были произведены в соответствии с законодательством Российской Федерации;
- предоставление уполномоченным федеральным органам исполнительной власти необходимой информации относительно предписаний, разрешений, аккредитации или лицензирования;
- выполнение в организации по взятию и проверке крови всех требований законодательства Российской Федерации.
- иметь высшее медицинское или биологическое образование;
- иметь стаж работы не менее двух лет в одной или нескольких организациях по взятию и проверке крови, осуществляющих забор и (или) испытания крови человека, ее компонентов, или их получение, хранение и распределение.
- Определение дано по Директиве ЕС 2002/98/ЕС (прим. разработчика страндарта).
1 Область применения
- Данное приложения распространяются на лекарственные средства, получаемые из донорской крови или плазмы, фракционированной в Российской Федерации или импортированной в Российскую Федерацию. Настоящее Приложение распространяется также на исходные материалы и сырье для таких лекарственных средств (например, донорскую плазму). Установленные настоящим Приложением требования применимы также к стабильным фракциям донорской крови или плазмы (например, альбумина), которые включают в медицинские изделия.
- Данное приложение устанавливает специальные требования GMP к производству, хранению и транспортированию донорской плазмы, используемой для фракционирования и для производства лекарственных средств, получаемых из донорской крови или плазмы.
- Данное приложение устанавливает специальные условия в случаях импорта исходных материалов из третьих стран, а также в случаях программ фракционирования по договору для третьих стран.
- Данное приложение не распространяется на компоненты крови, предназначенные для трансфузии.
2 Основные принципы
- Лекарственные средства, получаемые из донорской крови или плазмы (а также их активные фармацевтические субстанции, используемые как исходных материалы), должны соответствовать требованиям настоящего стандарта и регистрационному досье на лекарственное средство. Они рассматриваются как биологические лекарственные средства. Исходные материалы для них содержат биологические субстанции, такие как человеческие клетки или жидкости (включая кровь или плазму). Исходные материалы, вследствие биологической природы их происхождения, имеют характерные особенности. Например, исходные материалы могут быть загрязнены инфицирующими агентами, в особенности вирусами. Поэтому качество и безопасность таких лекарственных средств зависят от контроля исходных материалов и источника их происхождения, а также от дальнейших технологических процессов, включая проверку на инфекционные маркеры, удаление и инактивацию вирусов.
- Все фармацевтические субстанции, используемые как исходный материалы для лекарственных средств, должны отвечать требованиям настоящего стандарта. В отношении взятия и проверки исходных материалов, получаемых из донорской крови или плазмы, нужно выполнять следующие требования. Взятие и проверка должны проводиться в соответствии с системой обеспечения качества, требованиями нормативных правовых актов Российской Федерации и спецификациями. Следует выполнять установленные требования к прослеживаемости от донора до реципиента и в отношении уведомлений о побочных действиях и нежелательных реакциях. При этом необходимо руководствоваться Государственной фармакопеей Российской Федерации.
- Ввезенные из третьих стран исходные материалы для производства лекарственных средств из донорской крови или плазмы, если эти лекарственные средства предназначены для применения или реализации в Российской Федерации, должны отвечать нормам не ниже, чем действующие в Российской Федерации нормы в отношении систем качества для организаций по взятию и проверке крови. Также должны соблюдаться установленные нормативными правовыми актами Российской Федерации требования по прослеживаемости от донора до реципиента и в отношении уведомлений о побочных действиях и нежелательных реакциях и обеспечиваться соответствие установленными требованиям к крови и ее компонентам.
- При выполнении программ фракционирования по договору с третьими странами исходные материалы, ввезенные из других стран, должны соответствовать установленным нормативными правовыми актами Российской Федерации требованиям. Работы, проводимые в Российской Федерации, должны в полной мере соответствовать настоящему стандарту. Следует выполнять требования к системам качества для организаций по взятию и проверке крови. Также выполнять требования к прослеживаемости от донора до реципиента и в отношении уведомлений о побочных действиях и нежелательных реакциях и обеспечиваться соответствие действующим требованиям к крови и ее компонентам.
- Настоящий стандарт распространяются на все стадии после взятия и проверки крови (например, обработка, включая разделение, замораживание, хранение и транспортировка к производителю). Как правило, за выполнение этих требований несет ответственность уполномоченное лицо производителя. Если специфические этапы обработки предназначенной для фракционирования плазмы проводятся в организации по взятию и проверке крови, то в такой организации может быть специально назначено уполномоченное лицо, но его присутствие и ответственность могут не совпадать с присутствием и ответственностью ответственного лица. Для обеспечения того, что обязанности уполномоченного лица, предусмотренные нормативными правовыми актами Российской Федерации, выполняются надлежащим образом, предприятие по фракционированию (производитель лекарственных средств) должно заключить договор с организацией по взятию и проверке крови в соответствии с главой 7 Части I настоящего стандарта, куда рекомендуется включать обязанности и подробные требования для обеспечения качества. Ответственное лицо организации по взятию и проверке крови и уполномоченное лицо предприятия по фракционированию (производству лекарственных препаратов, см. п. 3.5 Части I настоящего стандарта) должны принимать участие в подготовке договора. Уполномоченное лицо должно обеспечить проведение аудитов по проверке того, что организация по взятию и проверке крови исполняет условия договора,
- Специальные требования к документации и другие мероприятия в отношении исходных материалов для получаемых из плазмы лекарственных средств указывают в основном досье на плазму.
3 Обеспечение качества
- Обеспечение качества распространяется на все стадии от отбора доноров до поставки готовой продукции. Следует выполнять установленные требования к прослеживаемости, включая начальный этап до поставки плазмы на предприятие по фракционированию, взятие и проверку донорской крови или плазмы, предназначенной для производства лекарственных средств.
- Сбор крови или плазмы, которые используются в качестве исходных материалов в производстве лекарственных средств, следует проводить в организациях по взятию и проверке крови, а проверку в лабораториях, которые применяют системы качества, отвечающие установленным требованиям, имеют разрешение, выданное в установленном порядке, и подлежат регулярному инспектированию в соответствии с законодательством. При наличии у производителя программ фракционирования по договорам для третьих стран он обязан уведомить об этом уполномоченный федеральный орган исполнительной власти.
- При ввозе плазмы из третьих стран она должна поставляться только утвержденными поставщиками (например, организациями по взятию и проверке крови, включая внешние склады). Такие поставщики должны быть указаны в спецификациях на исходные материалы, подготовлены предприятием по фракционированию или производителем, согласованы уполномоченным лицом предприятия по фракционированию, расположенного в Российской Федерации, и утверждены в установленном порядке. Оценка и выдача разрешения на использование плазмы (плазмы для фракционирования) как исходного материала осуществляется в соответствии с п. 6.8 Части I настоящего стандарта.
- Предприятие по фракционированию или производитель готовых лекарственных средств должны проводить оценку поставщиков, включая аудит в соответствии с утвержденными инструкциями. Следует проводить повторную оценку квалификацию поставщиков с установленной периодичностью и с учетом оценки рисков.
- Предприятие по фракционированию или производитель готовых лекарственных средств должны заключить договоры с организациями по взятию и проверке крови, которые являются поставщиками. В такие договоры рекомендуется включать, как минимум, следующие условия:
- определение обязанностей и ответственности;
- требования к системе качества и документации;
- критерии отбора доноров и проведение испытаний;
- требования к сепарации крови на компоненты крови и плазму;
- требования к замораживанию плазмы;
- хранение и транспортирование плазмы;
- прослеживаемость и информирование после сдачи и взятия крови (в том числе о побочных эффектах).
- Для планирования, оценки и документального оформления всех изменений, которые могут оказать влияние на качество и безопасность продукции или прослеживаемость, должна быть предусмотрена система контроля изменений. Следует оценивать возможное влияние предлагаемых изменений. Должна быть определена необходимость дополнительных испытаний или проведения аттестации, особенно на стадиях инактивации и удаления вирусов.
- Для минимизации рисков, связанных с инфицирующими агентами, в том числе новыми, следует предусмотреть меры безопасности с учетом анализа рисков, чтобы:
- учитывать все факторы, связанные со снижением количества вирусов и (или) испытаниями на инфицирующие агенты или их аналоги;
4 Прослеживаемость и мероприятия, проводимые после взятия крови
- Следует организовать систему контроля, позволяющую проследить каждую сданную дозу крови или плазмы от донора и процедуры ее взятия через организацию по взятию и проверке крови до серии лекарственного средства, а также в обратном направлении.
- Следует определить ответственность за прослеживаемость продукции (пропуски чего-либо должны быть исключены):
- Данные, необходимые для полной прослеживаемости, должны храниться не менее 30 лет, если иное не установлено иное.
- В договоры, согласно п. 3.5 данного приложения, между организациями по взятию и проверке крови (в том числе контрольными лабораториями) и предприятием по фракционированию или производителем, должны включать условия прослеживаемости и выполнения мероприятий после взятия крови по всей цепи от взятия плазмы до всех производителей, ответственных за выдачу разрешения на выпуск готовой продукции.
- Организации по взятию и проверке крови должны уведомлять предприятие по фракционированию или производителя о любом случае, который может повлиять на качество или безопасность продукции, а также сообщать другую важную информацию, полученную после приема донора или выдачи разрешения на выпуск плазмы, например, обратную информацию (информацию, полученную после взятия крови). Если предприятие по фракционированию или производитель находятся другой стране, то информацию следует сообщать производителю, находящемуся в Российской Федерации, ответственному за выдачу разрешения на выпуск лекарственных средств. В обоих случаях такая информация, если она имеет отношение к качеству и безопасности готовой продукции, должна быть доведена до сведения соответствующего надзорного органа.
- В случае если результатом инспектирования надзорным органом организации по взятию и проверке крови является отзыв лицензии, необходимо также направить уведомление в соответствии с 4.5 данного приложения.
- Инструкции должны регламентировать обращение информации, полученной после взятия крови. При этом должны быть учтены лицензионные требования и процедуры информирования надзорного органа. Мероприятия, проводимые после взятия крови, должны осуществляться в соответствии с действующими требованиями.
5 Помещения и оборудование
- В целях сведения к минимуму возможности микробного загрязнения или внесения постороннего материала в серию плазмы, оттаивание и объединение единиц плазмы должны выполняться в зонах не ниже D по приложению 1 к настоящему стандарту. Следует носить одежду в соответствии с классом чистоты, маски для лица и перчатки. Все другие открытые операции в ходе технологического процесса следует выполнять в условиях, соответствующих приложению 1 к настоящему стандарту.
- Следует регулярно контролировать параметры окружающей среды согласно приложению 1 к настоящему стандарту, особенно при открывании контейнеров с плазмой и в течение последующих оттаивании и объединении. Должны быть установлены допустимые значения для параметров.
- При производстве лекарственных препаратов, получаемых из донорской плазмы, следует предусматривать инактивацию или удаление вирусов и принимать меры по предотвращению перекрестного загрязнения обработанной продукции и необработанной продукцией. Для стадий технологического процесса, которые проводятся после инактивации вирусов необходимо использовать специально предназначенные выделенные помещения и оборудование.
- Не допускается проведение аттестации методов снижения числа (инактивации) вирусов в производственных помещениях и на оборудовании, с целью исключения их загрязнения вирусами, которые используются во время испытаний. Аттестацию следует проводить в соответствии с установленными требованиями.
6 Производство
Исходные материалы
- Исходные материалы должны отвечать требованиям Государственной фармакопеи Российской Федерации и условиям регистрационного досье, в том числе в основного досье плазмы. Эти условия включаются в договор между организацией по взятию и проверке крови и предприятием по фракционированию, либо производителем (п. 3.5 данного приложения), предусмотрев контроль в рамках системы качества.
- Исходное сырье для программ фракционирования по договору для третьих стран должно соответствовать требованиям п. 2.4 данного приложения.
- В зависимости от типа сбора (например, сбор цельной крови или автоматический аферез) могут потребоваться различные стадии обработки. Все стадии обработки, например, разделение (в том числе центрифугирование), отбор проб, маркировка, замораживание должны быть регламентированы инструкциями.
- Следует не допускать перепутывания единиц и образцов, особенно во время маркировки, а также загрязнения, например, при отрезании сегментов трубок или укупоривании контейнеров.
- Замораживание является критической стадией выделения белков, которые в плазме являются лабильными, например, факторов свертываемости. По этой причине замораживание должно осуществляться с помощью аттестованных методов как можно быстрее после взятия крови в соответствии с требованиями Государственной фармакопеи Российской Федерации.
- Условия хранения и транспортирования крови или плазмы к предприятию по фракционированию должны быть определены и оформлены документально на всех этапах цепи поставки. Следует извещать предприятие по фракционированию о любых отклонениях от установленной температуры. Следует использовать аттестованные (испытанные) методы и оборудование.
Оценка и выдача разрешения на выпуск плазмы для фракционирования, используемой как исходное сырье
- Разрешение на выпуск плазмы для фракционирования (например, из карантина) может дано только при выполнении условий системы качества (и инструкций), которые обеспечивают требуемое качество для производства готовой продукции. Плазма может быть поставлена предприятию по фракционированию или производителю только после документального подтверждения ответственным лицом (или в случае взятия крови или плазмы в третьих странах лицом с аналогичными обязанностями и квалификацией) того, что плазма для фракционирования соответствует требованиям и спецификациям, установленным в соответствующих договорах, а также того, что все стадии были выполнены в соответствии с настоящим стандартом.
- Уполномоченное лицо должно дать разрешение на использование всех контейнеров с плазмой для фракционирования при их поступлении на предприятие по фракционированию. Уполномоченное лицо должно подтвердить, что плазма соответствует всем требованиям Государственной фармакопеи Российской Федерации, удовлетворяет условиям регистрационного досье, в том числе основного досье плазмы, или в случае использования плазмы для программ фракционирования по договору для третьих стран всем требованиям, предусмотрены п. 2.4. данного приложения.
Обработка плазмы для фракционирования
- Стадии процесса фракционирования различаются в зависимости от продукции и производителя. Как правило, они включают различные операции фракционирования, а некоторые из них могут способствовать инактивации или удалению возможных загрязнений.
- Следует установить и выполнять требования к процессам объединения, отбора проб из объединенной плазмы, фракционирования и инактивации или удаления вирусов.
- При выполнении процесса инактивации вирусов следует использовать аттестванные методы, которые должны соответствовать методам, которые были использованы при исследовании инактивации вирусов. Необходимо проводить тщательное расследование всех неудавшихся процедур инактивации вирусов. Особо важно значение имеет выполнение аттестованного технологического процесса при снижении числа вирусов, поскольку какие-либо отклонения могут представлять риск для безопасности готовой продукции. Следует иметь инструкцию по анализу указанных рисков.
- Повторную обработку или переработку допускается выполнять только после анализа рисков для качества и только на определенных стадиях технологического процесса, что указывается в соответствующей документации.
- Следует организовать систему четкого разделения и (или) различения лекарственных средств или промежуточной продукции, которые прошли и которые не прошли процедуру снижения числа вирусов.
- Если на одной производственной площадке обрабатывают плазму или промежуточную продукцию различного происхождения, то возможна организация производства циклами при условии четкого разделение и наличия аттестованных методов очистки и в зависимости от результатов анализа рисков и в соответствии с действующими требованиями. Необходимость использования специализированного оборудования следует определить с учетом анализа рисков в случае реализации программ фракционирования по договору с третьими странами.
- Срок хранения промежуточной продукции, предназначенной для хранения, необходимо устанавливать на основе данных о стабильности.
- Должны быть установлены и документально оформлены требования к хранению и транспортированию промежуточной продукции и готовых лекарственных средств на всех этапах цепи поставки. Следует использовать аттестованные (испытанные) оборудование и методы.
7 Контроль качества
- Требования к испытаниям на вирусы или другие инфицирующие агенты должны устанавливаться с учетом новых знаний о них и наличия аттестованных методов испытаний.
- Следует контролировать первый однородный пул плазмы (например, после отделения криопреципитата от плазмы) с помощью аттестованных методов с требуемой чувствительностью и специфичностью согласно Государственной фармакопее Российской Федерации.
8 Выпуск промежуточной и готовой продукции
- Допускается выпуск только серий, которые получены из пулов плазмы, признанных по результатам контроля негативными в отношении маркеров вирусных инфекций, передаваемых через кровь, а также соответствующими требованиям Российской Федерации (включая специальные пределы, ограничивающие содержание вирусов) и утвержденным спецификациям (в частности, основному досье на плазму).
- Разрешение на выпуск промежуточной продукции, предназначенной для дальнейшей обработки внутри производственной площадки или поставки на другую производственную площадку, а также разрешение на выпуск готовых лекарственных средств выдается уполномоченным лицом с соблюдением установленных требований.
- Уполномоченное лицо должно выдавать разрешение на выпуск промежуточной или готовой продукции, используемой для программ фракционирования по договору для третьих стран, на основании нормативов, согласованных с заказчиком, а также в соответствии с требованиями настоящего стандарта. Если такие лекарственные препараты не предназначены для применения в Российской Федерации, к ним могут не применяться требования Государственной фармакопеи Российской Федерации.
9 Хранение образцов пулов плазмы
- Допускается использование одного пула плазмы для производства нескольких серий и (или) лекарственных средств. Контрольные образцы каждого пула плазмы, а также соответствующая документация должны храниться не менее одного года после окончания срока хранения полученного из этого пула лекарственного средства с наибольшим сроком хранения из всех лекарственных средств, полученных из этого пула плазмы.
10 Утилизация отходов
Организация должна разработать и внедрить инструкции по безопасному обращению, хранению и утилизации отходов, использованных и отклоненных материалов (например, загрязненных материалов, материалов от инфицированных доноров, а также крови, плазмы, промежуточной и готовой продукции с истекшими сроками годности).Перевод ООО «Чистые технологии», Федотов А. Е.
Корректировка 27.10.2023
Приложение 1 к правилам GMP EC Утверждено 22.08.2022
Производство стерильных лекарственных средств
Оглавление
|
Номер раздела |
Основное содержание |
|
1. Область применения |
Включает дополнительные области (иные, чем про- изводство стерильных лекарственных средств), в ко- торых могут применяться общие принципы данного приложения. |
|
2. Общие принципы |
Общие принципы, применяемые к производству сте- рильных лекарственных средств. |
|
3. Фармацевтическая си- стема качества (ФСК) |
Выделяет специальные требования ФСК к производ- ству стерильных лекарственных средств. |
|
4. Помещения |
Общее руководство, содержащее специальные требо- вания к проектированию помещений и аттестации, включая использование барьерной технологии. |
|
5. Оборудование |
Общие требования к конструкции и эксплуатации оборудования. |
|
6. Инженерные системы |
Специальные требования к таким инженерным си- стемам как подготовка воды, воздуха и вакуума. |
|
7. Персонал |
Требования к специальному обучению, знаниям и квалификации. Содержит также руководство по атте- стации персонала. |
|
8. Производство и специальные технологии |
Руководство по асептическим процессам и процессам с финишной стерилизацией. Руководство по стерили- зации продукции, оборудования и элементов упаков- ки. Рассматривает также разные процессы, такие как лиофилизация и «выдувание-наполнение- герметизация», к которым могут предъявляться спе- циальные требования. |
|
9. Контроль окружающей среды и процессов |
Данный раздел отличается от раздела 4 тем, что он относится к текущему контролю (мониторингу) с учетом проекта (конструкции) систем и установления уровней предупреждения и анализа тенденций Приводятся также требования к имитации асептиче- ских процессов. |
|
10. Контроль качества |
Руководство по ряду специфических требований к контролю качества стерильной продукции. |
|
11. Термины и определения |
Пояснения к специальным терминам. |
1.Область применения
Производство стерильных лекарственных средств включает в себя широкий спектр видов продукции (активные субстанции, вспомогательные вещества, первичные упако- вочные материалы, готовые дозированные формы), размеров серий (от одной единицы до многих единиц продукции), процессов (от высоко автоматизированных систем до ручных операций), упаковочных материалов и технологий (например, биотехнология, классиче- ское производство на основе малых молекул и закрытые системы). Настоящее приложе- ние дает общее руководство, которое рекомендуется использовать при проектировании и контроле помещений, оборудования, систем и процессов для производства всей стериль- ной продукции с применением анализа рисков для качества (АРК) для предотвращения загрязнения готовой продукции микроорганизмами, частицами и эндотоксинами (пироге- нами).
АРК применяется к данному документу в целом, как правило, без ссылок в отдель- ных разделах. В случаях, когда заданы пределы, периодичности или области изменения, их следует рассматривать как минимальные требования. Они установлены на основании имеющегося опыта нормативной практики с целью обеспечения безопасности пациентов.
Настоящее приложение дает руководство по производству стерильной продукции. Однако некоторые принципы и правила, такие как стратегия контроля загрязнений, проек- тирование помещений, классификация чистых помещений, аттестация (испытаний), теку- щий контроль (мониторинг) и порядок переодевания могут применяться и в производстве другой продукции, не являющейся стерильной, например, некоторые жидкости, кремы, мази и другая продукция с низким содержанием микробных загрязнений, но где важны контроль и снижение загрязнения микроорганизмами, частицами и эндотоксина- ми/пирогенами.
В случаях, когда производитель применяет данное руководство к производству не- стерильной продукции, ему следует четко указывать, какие принципы применяются с до- кументальным оформлением и показать соответствие этим принципам.
2.Основные принципы
2.1. К производству стерильной продукции предъявляются специальные требования с целью сведения к минимуму риска загрязнения микроорганизмами, частицами и эндоток- синами/пирогенами. К таким основным требованиям относятся: i) Проект (конструкция) зданий, помещений, оборудования и процессов и их атте- стация (испытания), где требуется, и последующий контроль следует выполнять в соот- ветствии с Правилами производства лекарственных средств (GMP). При применении тех- нологий (например, барьерных систем с ограниченным доступом – (RABS), изоляторов, роботизированных систем, ускоренных/альтернативных методов контроля) следует стре- миться к повышению защиты продукции от возможных внешних источников загрязнения эндотоксинами/пирогенами, частицами и микроорганизмами, таких как персонал, матери- ал, окружающая среда и быстрому обнаружению потенциальных загрязнений в окружаю- щей среде и продукции. ii)Персонал должен иметь необходимые квалификацию и опыт, пройти обучение и относиться к работе с особым вниманием в плане защиты стерильной продукции в про- цессе производства, упаковки и реализации. iii)Разработку, приемку, аттестацию и контроль процессов и систем мониторинга производства стерильной продукции должен выполнять персонал, имеющий знания в об- ласти технологии, инженерных систем и микробиологии. iv)Исходные и упаковочные материалы следует контролировать и испытывать в установленном порядке на предмет пригодности к использованию по загрязнению жизне- способными организмами и эндотоксинами/пирогенами. 2.2. Процессы, оборудование, помещения и производственная деятельность следует рассматривать в соответствии с принципами анализа рисков, чтобы обеспечивать преду- преждающие действия по идентификации, научной оценке и контролю потенциальных рисков для качества. При использовании альтернативных подходов их следует обосно- вать, выполнить оценку и обеспечить снижение рисков с учетом выполнения требований данного приложения. В первую очередь приоритетными задачами анализа рисков являются проектирова- ние зданий, помещений, оборудования и процессов в соответствии с заданными требова- ниями с последующим применением хорошо отработанных технологий и, наконец, при- менения систем текущего контроля (мониторинга) как средства подтверждения того, что проект и технологии правильно реализованы и работают в соответствии с требованиями. Контроль (мониторинг) и испытания сами по себе не обеспечивают гарантию стерильности. 2.3. Следует применять стратегию контроля загрязнений (СКЗ) на всем предприятии с целью выявления критических контрольных точек и оценки эффективности всех приня- тых решений (проекта, технологии, технических средств и организации производства) и методов контроля, применяемых для снижения риска для качества и безопасности про- дукции. Стратегия контроля загрязнений является комплексной мерой по надежному пре- дупреждению загрязнений. Следует критически пересматривать СКЗ и, если требуется, вносить в нее изменения имея в виду постоянное совершенствование методов производ- ства и контроля. Их эффективность является частью периодического пересмотра системы управления. При наличии действующих систем контроля и их использования, может не потребоваться их изменения, но это должно быть отмечено в СКЗ и взаимодействие меж- ду системами должно быть понятным. 2..4. Контроль загрязнений и меры, предпринимаемые для сведения к минимуму рис- ка загрязнений от источников микроорганизмов, эндотоксинов/пирогенов и частиц, вклю- чают ряд взаимосвязанных событий и действий. Как правило, они оцениваются и контро- лируются по отдельности, но учитывать их эффективность следует в комплексе. 2.5. Разработка СКЗ требует полного знания техники и технологии. Потенциальные источники загрязнений могут выделять микроорганизмы и клеточный дебрис (например, пирогены, эндотоксины) и частицы (например, стекло и другие видимые и невидимые частицы).В СКЗ должны быть учтены следующие элементы (перечень неисчерпывающий)
- проект предприятия и процессов, включая требуемую документацию;
- помещения и оборудование;
- персонал;
- инженерные системы;
- контроль исходных материалов, включая внутрипроизводственный контроль;
- первичные упаковочные и укупорочные материалы;
- утверждение поставщиков основных материалов, исполнителей работ по стери- лизации материалов и одноразовых систем, а также оказывающих основные услуги;
- управление работами, выполняемыми сторонними организациями, и контроль доступности информации, передаваемой сторонами, например, стерилизации по договору;
- оценка рисков для процесса;
- аттестация (испытания) процессов;
- аттестация процессов стерилизации;
- профилактическое обслуживание - техническое обслуживание оборудования, инженерных систем и помещений (плановое и внеплановое), выполняемое так, чтобы не увеличить риск загрязнений;
- очистка и дезинфекция;
- системы текущего контроля (мониторинга), включая оценку целесообразности применения научно обоснованных альтернативных методов оптимизации обнаружения загрязнений окружающей среды;
- порядок предупреждения – анализ тенденций, подробные расследования, уста- новление первопричин, корректирующие и предупреждающие действия (CAPA) и необ- ходимости применения более эффективных методов расследования;
- постоянное улучшение, основанное на информации о перечисленных выше факторах.
3. Фармацевтическая система качества (ФСК)
3.1. Производство стерильной продукции представляет собой комплекс действий, ко- торые требуют дополнительного контроля и мер по обеспечению качества выпускаемой продукции. В соответствии с этим ФСК производителя должна быть ориентирована на выполнение специальных требований к производству стерильной продукции и обеспечи- вать эффективный контроль всех операций так, чтобы свести к минимуму риск загрязне- ния стерильной продукции микроорганизмами, частицами и эндотоксинами/пирогенами. В дополнение к требованиям к фармацевтической системе качества, приведенных в главе 1 правил GMP (часть I – Основные требования к лекарственным средствам) ФСК в произ- водстве стерильной продукции должна включать в себя следующее:- Эффективная система анализа рисков должна быть встроена в жизненный цикл про- дукции с целью сведения к минимуму микробного загрязнения для обеспечения качества производимой стерильной продукции.
- Производитель должен иметь достаточные знания и опыт работы в отношении выпускаемой продукции, оборудования, инженерных систем и технологии производства, имеющих влияние на качество продукции.
- Анализ основной (первичной) причины сбоев в работе методов, процессов или отказов оборудования является ключевым моментом для правильного понимания риска для продукции и применения необходимых корректирующих и предупреждающих мер;
- Анализ рисков выполняется при разработке и функционировании СКЗ для иден- тификации, оценки, устранения (где требуется) и контроля риска загрязнений для преду- преждения, контроля и контроля риска загрязнений. Анализ рисков следует выполнять в документальной форме с обоснованием решений по снижению риска и приемлемости остаточного риска.
- Руководству предприятия следует тщательно рассматривать состояние с контро- лем на производстве в течение всего периода его работы. Следует регулярно рассматри- вать результаты анализа рисков при текущем контроле качества, контроле изменений и периодическом анализе качества.
- Завершающие операции, хранение и транспортирование стерильной продукции не должны подвергать риску готовую стерильную продукцию. Следует обращать внима- ние на целостность упаковки, риски загрязнения и предотвращение деградации за счет выполнения установленных требований к хранению продукции и обращению с нею.
- Лица, ответственные за сертификацию/выпуск стерильной продукции должны иметь доступ к информации о производстве и качестве и обладать необходимыми знания- ми и опытом в области производства стерильной продукции и ее критических для каче- ства свойствах. Это необходимо, чтобы указанные лица могли установить, что стерильная продукция произведена в соответствии с требованиями, утвержденными процессами и имеет требуемое качество
4.Помещения
4.1. Производство стерильной продукции следует организовывать в требуемых чистых помещениях, вход в которые должен осуществляться через комнаты переодевания, которые выполнены как воздушные шлюзы для персонала, и воздушные шлюзы для оборудования и материалов. Чистые помещения и комнаты переодевания должны быть выполнены согласно требованиям стандартов на чистые помещения с подачей приточного воздуха через фильтры требуемой эффективности. Текущий контроль (мониторинг) должен быть научно обоснован. Он должен давать эффективную оценку состояния окружающей среды в чистых помещениях, воздушных шлюзах и передаточных камерах. 4.2. Подготовка исходных материалов, приготовление продукции и наполнение должны выполняться с требуемым разделением техническими и организационными мера- ми в чистом помещении или в других помещениях для предотвращения перепутывания и загрязнения. 4.3. Системы с ограниченным доступом (RABS) или изоляторы являются эффектив- ными средствами в обеспечении требуемых условий и сведении к минимуму микробного загрязнения, связанного с прямыми вмешательствами человека в критической зоне. Их применение следует учесть в СКЗ. В случае применения любых подходов, отличающихся от RABS или изоляторов, их следует обосновать. 4.4. Для производства стерильных лекарственных средств определены четыре типа чистых помещений (зон).Зона А: Локальная зона для выполнения операций с высоким уровнем риска (напри- мер, асептическая технологическая линия; зона наполнения; бункер с пробками для уку- порки; зоны, где первичные упаковочные материалы или материалы для соединений в асептических условиях находятся в открытом состоянии под защитой непосредственно поступающего чистого воздуха). Как правило, эти условия обеспечиваются локальной за- щитой потоком воздуха, такой как рабочие зоны с однонаправленным потоком воздуха в RABS или изоляторах. Поддержание однонаправленного потока воздуха должно быть по- казано и подтверждено при аттестации для всей зоны А. Непосредственное вмешательство оператора в зону А (например, при отсутствии защитного барьера и перчаточных боксов) должно быть сведено к минимуму при разработке проекта помещений, конструкции обо- рудования и разработки процесса.
Зона В: В процессах асептического приготовления и наполнения – зона чистого по- мещения, непосредственно окружающая зону А (если нет изолятора). Следует непрерывно контролировать перепад давления воздуха. При использовании изолятора могут преду- сматриваться чистые помещения низшего класса, чем зона В (п. 4.20).
Зоны С и D: Чистые зоны для выполнения менее критических стадий производства стерильной продукции с наполнением в асептических условиях или в качестве окружаю- щей среды для изоляторов. Они могут также использоваться для приготовления/наполнения продукции, подлежащей финишной стерилизации (раздел 8, специфические особенности операций при финишной стерилизации).
4.5. Все критические поверхности в чистых помещениях и критических зонах долж- ны быть гладкими, непроницаемыми и не иметь повреждений с целью сведения к мини- муму выделения или накопления частиц или микроорганизмов. 4.6. Чтобы уменьшить накопление пыли и облегчить очистку, в помещении не долж- но быть труднодоступных для очистки мест, поэтому выступающие части, полки, шкафы и оборудования должны быть минимальными. Конструкция дверей должна предусматри- вать отсутствие пазов, которые невозможно очистить. По этой причине применение раз- движных дверей может быть нежелательным. 4.7. Выделение частиц материалами конструкций помещения и используемых в нем, должно быть минимальным. Материалы должны допускать многократную обработку мо- ющих, дезинфицирующих и спороцидных средств. 4.8. Конструкция потолков и их герметизация должны предотвращать проникание за- грязнений из пространства над ними. 4.9. Не допускается устанавливать раковины и сливы в зонах А и В. В других зонах следует предусматривать разрыв струи между оборудованием или раковиной и канализа- ционной трубой (воронкой). При удалении стоков в чистых помещениях более низких классов следует предусматривать трапы или гидрозатворы для предотвращения обратного потока, которые следует регулярно очищать, дезинфицировать и обслуживать. 4.10. Перемещение оборудования и материалов в чистые помещения и из них являет- ся одной из основных причин загрязнения. Следует оценивать любые действия персонала, которые могут нарушить чистоту чистых помещений или критических зон. Если их нельзя исключить, то следует предусмотреть необходимые меры контроля. 4.11. Следует перемещать материалы, оборудование и компоненты в зоны А и В в одном направлении. Где это возможно, предметы следует стерилизовать и перемещать в эти зоны через проходные стерилизаторы (например, двухдверные автоклавы или шкафы (туннели) депирогенизации, герметично встроенные в стены). Если стерилизация после перемещения невозможна, следует применить и аттестовать процессы, исключающие по- падание загрязнений (например, путем эффективной дезинфекции при перемещении, си- стемы быстрого перемещения для изоляторов или удерживающие бактерии фильтры для жидких и газообразных сред). Удаление предметов из зон А и В (например, материалов, отходов, проб окружающей среды) следует выполнять по отдельному маршруту в одном направлении. Если это невозможно, то допускается перемещение материалов внутрь и наружу с разделением во времени и организацией контроля для исключения возможного загрязнения перемещаемых внутрь предметов. 4.12. Конструкция и порядок использования воздушных шлюзов должны обеспечи- вать физическое разделение, чтобы свести к минимуму загрязнения разных зон микроор- ганизмами и частицами. Воздушные шлюзы предназначены для движения персонала и ма- териалов между зонами разных типов. Как правило, предусматриваются отдельные воз- душные шлюзы для персонала и материалов. Если это нецелесообразно, то следует преду- сматривать разделение во времени движения персонала и материалов. В воздушных шлю- зах следует предусматривать эффективное обтекание воздухом, прошедшим фильтрацию, для того, чтобы поддерживать в нем заданный класс чистоты. Зона перед выходом из воз- душного шлюза должна иметь в оснащенном состоянии тот же уровень чистоты (по ча- стицам и микроорганизмам), что и чистое помещение, в которое она ведет. Рекомендуется предусматривать отдельные комнаты переодевания для входа в зоны В и выхода из них. Если это нецелесообразно, то следует предусмотреть разделение входа и выхода во вре- мени по инструкции. Если согласно СКЗ риск загрязнений высок, то следует предусматри- вать отдельные комнаты переодевания для входа в производственные зоны и выхода из них.Конструкция воздушных шлюзов должна удовлетворять следующим требованиям:
- Воздушные шлюзы для персонала:
Предусматриваются зоны с повышением уровня чистоты (например, из зоны D в зо- ну С, затем в зону В). Как правило, мойки для рук должны устанавливаться в первой части комнаты переодевания и не должны находиться в комнате переодевания, непосредственно ведущей в зону В.
2. Материальные воздушные шлюзы (для перемещения материалов и оборудования):
-
- Допускается перемещение через материальные шлюзы и передаточные каме- ры в зоны А и В только материалов и оборудования, включенных в утвержденный перечень после аттестации этого процесса. Оборудование и матери- алы (предназначенные для использования в зонах А) следует защитить при перемещении через зоны В. Следует получить разрешение на перемещение любых предметов, не включенные в утвержденный перечень, в качестве ис- ключения. Следует выполнить оценку риска и принять меры по его снижению с документальным оформлением согласно СКЗ производителя и предусмотреть специальную программу дезинфекции и контроля, согласованную служ- бой обеспечения качества.
-
- Конструкция передаточных камер должна предусматривать защиту среды с более высоким уровнем чистоты, например, за счет эффективного обдувания воздухом от приточного фильтра.
- При перемещении материалов или оборудования из чистого помещения с меньшими требованиями к чистоте или из неклассифицированной зоны в зону с более высокими требованиями к чистоте следует выполнять их очистки и дезинфекцию с учетом риска согласно СКЗ.
Барьерные технологии
4.18. Изоляторы или RABS позволяют применять технологии, отличающиеся от обычных процессов. Их конструкция должна обеспечивать защиту путем отделения зоны А от окружающего помещения. Следует свести к минимуму опасности при передаче или удалении предметов в ходе процесса с применением передовых технологий передачи или аттестованных систем, которые надежно предотвращают загрязнение и совместимы с применяемой технологией. 4.19. Технические и технологические решения должны обеспечивать поддержание требуемых условий для защиты открытого продукта при выполнении операций.- Изоляторы:
- Конструкция открытых изоляторов должна включать зону А с непосредственной подачей воздуха в критическую зону и однонаправленным потоком, который обтекает от- крытый продукт и удаляется от него во время процесса.
- Конструкция закрытых изоляторов должна включать зону А и необходимую за- щиту продукта в ходе процесса. В закрытых изоляторах поток воздуха может не быть полностью однонаправленным, если в них выполняются простые операции. В то же время никакие турбулентные потоки не должны приводить к риску загрязнения открытого про- дукта. Если технологическая линия входит в состав закрытого изолятора, то он должен включать зону А с непосредственной подачей воздуха в критическую зону и однонаправ- ленным потоком, который обтекает открытый продукт и удаляется от него во время про- цесса.
- Изоляторы с пониженным давлением только в случаях, когда изолирование про- дукта играет важную роль (например, радиофармацевтические препараты). Следует при- нять специальные меры по контролю риска для защиты критической зоны.
- RABS
Конструкция систем RABS должна обеспечивать выполнение требований к зоне А за счет однонаправленного потока воздуха и защитой критической зоны непосредственно поступающим в нее воздухом. Поток воздуха должен быть направлен от критической зо-
Конструкция систем RABS должна обеспечивать выполнение требований к зоне А за счет однонаправленного потока воздуха и защитой критической зоны непосредственно поступающим в нее воздухом. Поток воздуха должен быть направлен от критической зоны в окружающую среду.
4.20. Среда, окружающая изолятор или RABS, должна обеспечивать сведение риска переноса загрязнений к минимуму.- Изоляторы:
- Открытый изолятор следует устанавливать, как правило в зоне С (как минимум). Закрытый изолятор следует устанавливать, как правило, в зоне D (как минимум). Решение о классе окружающей среды должно быть основано на оценке риска и включено в СКЗ.
- При оценке риска, связанного с изолятором, для СКЗ нужно учесть следующие ключевые факторы (перечень неисчерпывающий): программу борьбы с биозагрязнениями; степень автоматизации; влияние манипуляций в перчатках, которые потенциально могут нарушить непосредственную защиту точек критического процесса, влияние возможного нарушения целостности барьера/перчаток, применяемый порядок передачи и такие дей- ствия как наладка или техническое обслуживание, при которых может потребоваться от- крывание дверей до окончания биологической обработки изолятора. При обнаружении дополнительных факторов риска следует повысить требования к окружающему помеще- нию, если иное не обосновано в СКЗ.
- Для открытых изоляторов следует выполнить анализ потоков воздуха в отверстиях и показать отсутствие проникания воздуха внутрь.
- RABS
Для асептических процессов система RABS должна быть установлена, как минимум в зоне В. Следует выполнить анализ потоков воздуха и показать отсутствие проникания воздуха внутрь, включая дверные проемы, если это применимо.
4.21. Следует подтвердить достаточную механическую и химическую прочность ма- териалов перчаточных систем (как изоляторов, так и RABS). Периодичность замены пер- чаток должна быть определена в СКЗ.- Изоляторы:
- Следует выполнить испытания на утечку (контроль целостности) перчаточных си- стем в изоляторах обоснованным для этой цели методом с учетом фактора критичности. Испытания на утечку должны проводиться с установленной периодичностью. Как прави- ло, контроль целостности перчаток следует выполнять с минимальной частотой в начале и в конце производства каждой серии или цикла (кампании). Дополнительный контроль це- лостности может потребоваться в зависимости от аттестованной длительности цикла. Текущий контроль целостности перчаток должен включать визуальный осмотр при каждом использовании и выполняться после выполнения любой операции, которая может повлиять на целостность системы
- Контроль целостности (утечек) изолирующей системы следует выполнять с опре- деленной периодичностью.
- RABS
Перчатки для систем RABS с зонами А следует стерилизовать перед установкой и стерилизовать или обрабатывать эффективным аттестованным методом перед каждым производственным циклом. В случае контакта внутреннего объема с окружающей средой при выполнении операций следует провести дезинфекцию аттестованным методом после каждого случая контакта. Перчатки следует проверять визуально при каждом использова- нии и выполнять контроль целостности с установленной периодичностью (интервалами).
4.22. Следует определить и контролировать применение методов обработки (очистки и дезинфекции и, где требуется инактивации биологических материалов). Очистка являет- ся важным этапом перед проведением дезинфекции. Любые остатки после проведения очистки могут снизить эффективность обработки. Следует показать, что средства для очистки и дезинфекции не оказывают вредного влияния на продукцию, производимую в RABS или изоляторах.- Изоляторы
Процесс обработки внутри изолятора следует выполнять автоматическими метода- ми, аттестовать и контролировать для заданных параметров цикла. Он должен предусмат- ривать применение спороцидных средств в пригодной форме (например, газообразной или в виде паров). Перчатки должны быть растянуты и пальцы разделены, чтобы обеспечить контакт со средством. Используемые методы очистки и спороцидной дезинфекции долж- ны обеспечивать отсутствие жизнеспособных микроорганизмов на внутренних поверхно- стях и в критической зоне изолятора.
2. При спороцидной дезинфекции следует постоянно применять спороцидные сред- ства по аттестованным методам, для которых показано, что обработка проводится надеж- но для всей внутренней поверхности и обеспечивает требуемую окружающую среду для асептических процессов.Аттестация чистых помещений и установок для очистки воздуха
4.23. Чистые помещения и установки для очистки воздуха, такие как боксы с однона- правленным потоком воздуха, RABS и изоляторы, используемые в производстве стериль- ной продукции, должны быть аттестованы (испытаны) на соответствие требуемым показа- телям окружающей среды. Каждая производственная операция требует определенной чи- стоты окружающей среды в эксплуатируемом состоянии для сведения риска загрязнения продукции и материалов к минимуму. Следует поддерживать определенные уровни чи- стоты в оснащенном и в эксплуатируемом состояниях. 4.24. Аттестацию чистых помещений и установок для очистки воздуха следует проводить, используя методологию приложения 15. Следует четко различать аттестацию чистых помещений (включая классификацию) от текущего контроля в эксплуатации. 4.25. Аттестация чистых помещений и установок для очистки воздуха, является всеохватывающим процессом оценки их своему назначению. Аттестация чистых помеще- ний и установок для очистки воздуха, будучи частью требований приложения 15, должна включать (где это нужно по условиям проекта или их эксплуатации) испытания:- Установленнойсистемыфильтровнаутечкуи целостность;
- Потоковвоздуха:расходаи скорости;
- Перепадовдавлениявоздуха;
- Направления потоков воздуха и визуализацию;
- Микробного загрязнения воздуха и поверхностей;
- Температуры;
- Относительной влажности;
- Восстановления чистоты;
- Герметизации (утечек)
Аттестация чистых помещений и установок для очистки воздуха выполняется по серии стандартов ИСО 14644.
4.26. Классификация чистых помещений входит в состав работ по их аттестации и является методом оценки соответствия общей чистоты воздуха чистых помещений и уста- новок для очистки воздуха заданным требованиям путем определения концентрации ча- стиц. Работы по классификации следует планировать и выполнять так, чтобы не оказать влияния на процесс качество или качество продукции. Например, первоначальную классификацию следует выполнять, а повторная классификация - имитации операций или имитации асептического процесса. 4.27. При классификации чистых помещений следует определять общее число частиц с размерами равными и большими 0.5 и 5,0 мкм. Испытания следует выполнять как в оснащенном, так и в эксплуатируемом состояниях на соответствие пределам, указанным в таблице 1.Таблица 1 Максимальные концентрации частиц для целей классификации
|
Зона |
Пределы концентраций частиц с размерами ≥ 0,5 мкм/м3 |
Пределы концентраций частиц с размерами ≥ 5,0 мкм/м3 |
||
|
Оснащенное состояние |
Эксплуатируемое состояние |
Оснащенное состояние |
Эксплуатируемое состояние |
|
|
A |
3 520 |
3 520а |
Не заданоa) |
Не заданоa) |
|
B |
3 520 |
352 000 |
Не заданоa) |
2 930 |
|
C |
352 000 |
3 520 000 |
2 930 |
29 300 |
|
D |
3 520 000 |
Не заданоb) |
29 300 |
Не заданоb) |
a) Может выполняться классификация, включая частицы 5,0 мкм, если это указано в СКЗ или следует из тенденций предыдущего периода.
b) Пределы для зон D в эксплуатации не заданы. Производителю следует задать пре- делы в эксплуатации на основе оценка рисков или текущих данных, если требуется.
4.28. Порядок определения минимального числа точек отбора проб и их расположе- ния при классификации приведен в ИСО 14644-1. Для зон с асептическими процессами и окружающих их помещений (зоны А и В соответственно) могут предусматриваться дополнительные точки отбора проб и следует оценить все критические технологические зо- ны, такие как наполнения и герметизации вблизи питающих бункеров. Критические тех- нологические точки должны быть оценены исходя из риска с документальным оформле- нием и знаний о процессе и операциях, выполняемых в этой зоне. 4.29. Классификацию чистых помещений следует выполнять в оснащенном состоя- нии и в эксплуатации.- В оснащенном состоянии монтаж всех инженерных систем завершен, включая функционирование систем вентиляции и кондиционирования воздуха, когда основное технологическое оборудование установлено как предусмотрено, но не работает и персо- нал в помещении отсутствует.
- В эксплуатируемом состоянии монтаж чистого помещения завершен, системы вентиляции и кондиционирования воздуха работают полностью, оборудование установле- но и работает, как предусмотрено производителем с максимальным числом присутствую- щего персонала, который выполняет или имитирует выполнение технологических опера- ций.
- Соответствие пределов для общего числа частиц в оснащенном состоянии по таб- лице быть достигнуто после периода «очистки» по завершению работы и приведения в порядок (очистки) технологической линии. Период «очистки» (рекомендуемое значение – менее 20 мин.) следует определять при аттестации помещений и указывать в инструкциях по восстановлению аттестованного состояния после нарушения уровня чистоты при работе
Таблица 2
Максимально допустимые уровни микробного загрязнения при аттестации
|
Зона |
Проба воздуха, КОЕ/м3 |
Седиментационные пла- стины (диаметр 90 мм), КОЕ/4 ча) |
Контактные пластины (диаметр 55 мм), КОЕ/пластина |
|
A |
Отсутствие роста |
||
|
B |
10 |
5 |
5 |
|
C |
100 |
50 |
25 |
|
D |
200 |
100 |
50 |
а) Седиментационные пластины следует экспонировать в течение выполнения опе- раций и заменять, как требуется, но не более чем через 4 ч. Время экспозиции следует определять на основе анализа роста микрофлоры , причем оно не должно приводить к высыханию среды.
Примечание 1: Все методы, приведенные для аттестации отдельной зоны, должны применяться именно для этой зоны. Если один из методов таблицы не используется, или применяется альтернативный метод, то этому следует дать обоснование.
Примечание 2: Во всем документе используются пределы, выраженные в КОЕ. Если используются другие или новые методы, которые дают результаты, отличающиеся от КОЕ, то производитель должен научно обосновать эти пределы и, если возможно, сопо- ставить их с КОЕ.
Примечание 3: При аттестации процесса переодевания персонала следует применять пределы для контактных пластин и отпечаток с перчаток по таблице 6.
Примечание 4: Методы отбора проб не должны приводить к риску загрязнения про- изводственных операций.
4.32. Повторную аттестацию чистых помещений и установок для очистки воздуха следует выполнять периодически по утвержденным методикам. При повторной аттеста- ции следует проверять, как минимум:-
-
- класс чистоты (по общей концентрации частиц);
- целостность финишных фильтров;
- расходы воздуха;
- перепады давления воздуха между помещениями;
- скорость потока воздуха (примечание: для зон В; С и D контроль скорости воздуха следует выполнять в соответствии с оценкой риска документально оформленной в СКЗ. Но ее нужно проверять для линий наполнения, находящихся в зоне однонаправленного потока (например, при наполнении продукции, подлежащей финишной стерилизации или окружении зоны А и RABS). Для зон с неоднонаправленным потоком измерения скорости воздуха следует заменить на контроль восстановления.
-
Повторную аттестацию зон А и В следует проводить не реже, чем один раз в 6 мес. Повторную аттестацию зон С и D следует проводить не реже, чем один раз в 12 мес.
В состав работ по повторной аттестации должны входить, как минимум, перечис- ленные выше работы. Ее следует также проводить после устранения причин несоответ- ствия оборудования требованиям или условий в помещениях или после внесения измене- ний в оборудование, помещения или процессы. Следует определить значимость измене- ния в процессе их внесения. Примеры изменений, которые следует принять во внимание (перечень неисчерпывающий):
- Прерывание движения воздуха, которое может оказать влияние на работу помещения.
- Изменение проекта чистого помещения или эксплуатационных параметров системы отопления, вентиляции и кондиционирования.
- Работы по специальному техническому обслуживанию, которые оказывают влия- ние на работу помещения (например, замена финишных фильтров).
Дезинфекция
4.33. Дезинфекция чистых зон играет особенно важную роль. Их следует тщательно очищать и дезинфицировать в соответствии с письменной инструкцией. Для того, чтобы дезинфекция была эффективной, сначала необходимо выполнить очистку для удаления загрязнений с поверхностей. Процесс очистки должен эффективно удалять остатки дезин- фицирующих средств. Следует использовать более одного типа дезинфицирующего сред- ства, причем разные средства должны различаться по действию, и их совместное исполь- зование эффективно против бактерий и грибов. Следует периодически применять споро- цидное средство. Следует показать, что дезинфицирующие средства эффективны в тече- ние всего срока годности их использования с учетом соответствующего времени контакта, способа применения и поверхностей, на которых они используются. Следует регулярно проводить мониторинг, чтобы показать эффективность дезинфекции и обнаружить изме- нения в характере микрофлоры (например, организмов, устойчивых к применяемым в данное время дезинфицирующим средствам). 4,34. Процесс дезинфекции должен быть аттестован. При аттестации следует показать пригодность и эффективность дезинфицирующего средства при работе по принятому методу и на применяемом материале или материале-представителе, если это обосновано, а также подтвердить срок годности приготавливаемого раствора. 4.35. Дезинфицирующие и моющие средства для зон А и В должны быть стерильными перед применением. К дезинфицирующим средствам для зон С и D также может предъявляться требование стерильности, если указано в СКЗ. Если растворе- ние/приготовление дезинфицирующих и моющих средств выполняет производитель стерильной продукции, то следует принять меры защиты от загрязнений и контролировать уровень микробного загрязнения. Растворы следует хранить в вымытых сосудах (и, если требуется, стерилизованных) в течение определенного периода. Если применяются готовые дезинфицирующие и моющие средства, то могут быть признаны сертификаты анализа или соответствия в комплексе мер по аттестации поставщика. 4.36. При применении фумигации или дезинфекции парами (например, парами пере- киси водорода) для обработки чистых помещений и поверхностей, то следует понять и ат- тестовать эффективность каждого средства для фумигации и системы распыления.5.Оборудование
5.1. Следует иметь подробное и оформленное документально описание конструкции оборудования (включая чертежи и технологические схемы, при необходимости). Это должно входить в состав документации при первичной аттестации и в дальнейшем актуа- лизироваться. 5.2. Требования к контролю оборудования должны быть определены в спецификации пользователя на ранних стадиях разработки и подтверждаться при аттестации. Следует рассматривать случаи появления сигналов тревоги в ходе процесса и в оборудовании и проводить анализ тенденций. Периодичность оценки уровней тревоги зависит от критич- ности параметров (действия при тревоге в критических случаях следует предпринимать немедленно). 5.3. Конструкция, установка и расположение оборудования, фитингов (мест соедине- ния) и зон обслуживания должны, по возможности, предусматривать возможность работы с оборудованием, его технического обслуживания и ремонта снаружи чистой зоны. Если при выполнении технического обслуживания в чистом помещении поддерживать требуе- мый уровень чистоты и/или асептики невозможно, то следует принять меры предосто- рожности, такие как ограничение доступа в рабочую зону и допуск в нее только специаль- ного персонала, разработка четкой инструкции по работе и техническому обслуживанию. Следует проводить дополнительную дезинфекцию или контроль окружающей среды. При необходимости проведения стерилизации ее следует выполнять после максимально пол- ной разборки оборудования (насколько это возможно). 5.4.Следуетаттестоватьпроцессочисткиипоказать,чтоон:- Удаляет любые остатки или дебрис, которые могут оказать отрицательное влияние на эффективность используемого стерилизующего средства.
- Сводит к минимуму загрязнение продукции химическими веществами, микроор- ганизмами и частицами в течение выполнения процесса и до проведения дезинфекции.
6. Инженерные системы
6.1. Вид и объем контроля инженерных систем должен соответствовать риску, свя- занному с этими системами. Влияние систем должно быть установлено путем оценки рис- ка и оформлено документально как часть СКЗ. 6.2.Какправило,кфакторамповышенногорискавинженерныхсистемах относятся:- Прямой контакт с продуктом, например, воды для мойки или ополаскивания, газов и пара для стерилизации;
- Контакт с материалами, которые станут частью продукта;
- Контакт поверхностей, которые будут вступать в соприкосновение продуктом; iv Другие факторы, которые оказывают прямое влияние на продукт.
- Направление потока в трубопроводах, уклоны труб, их диаметры и длины.
- Данные о резервуарах и сосудах.
- Данные о вентилях, фильтрах сливах, точках отбора проб и потребления.
Системы подготовки воды
6.7. Системы подготовки и распределения воды должны быть разработаны, изготов- лены, установлены, приняты, аттестованы, их следует контролировать и обслуживать для предотвращения микробных загрязнений так, чтобы обеспечить надежную подготовку во- ды требуемого качества. Следует сводить к минимуму риск наличия частиц, загрязнения микроорганизмами и их роста, а также эндотоксинов/пирогенов (например, за счет укло- нов трубопроводов для обеспечения полного дренажа и исключения застойных зон). Сле- дует уделить особое внимание контролю и техническому обслуживанию фильтров (при их наличии). Приготовление воды должно выполняться в соответствии с действующей фар- макопеей. 6.8. Системы подготовки воды должны быть аттестованы с учетом сезонных измене- ний, чтобы обеспечить требуемый уровень контроля по физическим, химическим и мик- робиологическим показателям. 6.9. В трубопроводах должен поддерживаться турбулентный поток воды, чтобы све- сти к минимуму риск адгезии микроорганизмов и последующего образования биопленок. 6.10. Воду для инъекций (ВДИ) следует получать из воды, отвечающей требованиям, которые были получены при аттестации. Ее следует хранить и распределять так, чтобы исключить рост микроорганизмов (например, за счет постоянной циркуляции воды при температуре выше 70 °С). ВДИ следует приготавливать методом дистилляции или с при- менением другого процесса очистки, который эквивалентен дистилляции. Этот процесс может включать обратный осмос в сочетании с такими методами как электродеионизация, ультрафильтрация или нанофильтрация. 6.11. Если накопительные емкости для ВДИ оснащены гидрофобными вентиляцион- ными фильтрами, удерживающими бактерии, то эти фильтры не должны являться источ- никами загрязнения и их целостность должна проверяться до установки и после использо- вания. Следует предусмотреть меры по предотвращению образования конденсата на фильтре (например, за счет подогрева). 6.12. Для предотвращения образования биопленок следует проводить стерилизацию, дезинфекцию или регенерацию систем подготовки воды по утвержденному графику и при принятии мер при превышении допустимых пределов или требований спецификации. По- сле дезинфекции системы подготовки воды ее следует промыть аттестованным методом. После дезинфекции (регенерации) следует выполнить анализ воды. Результаты химиче- ского анализа должны быть утверждены до возобновления использования системы и должно быть проверено соответствие результатов микробиологического контроля (кон- троля эндотоксинов) требованиям спецификации, которые должны быть утверждены до сертификации (выпуска в реализацию) серии, для которой использована воды из системы. 6.13. Следует проводить регулярный химический и микробиологический анализ си- стем подготовки воды для подтверждения соответствия требованиям. Уровни предупре- ждения должны быть основаны на данных первоначальной аттестации и затем следует про- водить их периодическую оценку на основе данных повторной аттестации, текущего кон- троля и анализа. Этот анализ позволяет обнаружить отрицательные тенденции в работе си- стемы. Пробы должны отбираться во всех точках подключения и потребления с заданной периодичностью. Отбор проб должен отражать требования СКЗ и включать все точки по- требления, с заданными интервалами, чтобы обеспечить представительность проб воды на регулярной основе. Планы отбора проб должны быть основаны на данных аттестации и учитывать возможные наихудшие точки отбора проб и предусматривать отбор не менее од- ной представительной пробы каждый день для воды, используемой в производстве. 6.14. Случаи выхода за пределы предупреждения следует оформлять документально, рассматривать и анализировать на предмет установления, является ли этот случай един- ственным или результаты анализа указывают на отрицательную тенденцию или деграда- цию системы. Следует анализировать каждый случай выхода за предел действия, чтобы определить возможную первопричину и возможное влияние использования воды на каче- ство продукции и процесс производства. 6.15. В системах подготовки ВДИ следует предусматривать непрерывный контроль таких параметров как общий органический углерод (Total Organic Carbon, TOC) и прово- димость, поскольку они дают лучшую оценку работы всей системы, чем отдельные отбо- ры проб. Места размещения сенсоров следует определять на основе оценки риска.Пар, используемый для стерилизации
6.16. Исходная вода, подаваемая в генератор чистого пара, должна пройти необходи- мую очистку. Качество пара в соответствии с заданными пределами химических загрязне- ний и эндотоксинов должно обеспечиваться конструкцией генератора чистого пара, под- тверждено при аттестации и поддерживаться в эксплуатации. 6.17. Пар, используемый непосредственно для стерилизации, должен иметь требуе- мое качество и не должен содержать загрязнений на уровне, при котором может произой- ти загрязнение продукции или оборудования. Если генератор производит чистый пар, ис- пользуемый для непосредственной стерилизации материалов или поверхностей, вступаю- щих в контакт с продуктом (например, загрузки автоклава твердыми пористыми материа- лами), конденсат пара должен соответствовать действующей статье фармакопеи на ВДИ (микробиологический контроль конденсата пара не обязателен). Следует иметь график плана отбора проб чистого пара в представительном объеме для проведения регулярного анализа. Следует периодически оценивать другие параметры чистого пара, используемого для стерилизации на соответствие значениям, полученным при аттестации. В число этих параметров следует включать, например, неконденсируемые газы, степень сухости пара (сухости фракции), качество перегретого пара и конденсата, если не установлено иное.Системы сжатых газов и вакуума
6.18.Сжатые газы, которые могут вступать в прямой контакт с продуктом или по- верхностями первичной упаковки, должны иметь требуемую химическую и микробиоло- гическую чистоту и чистоту по частицам. Следует задать значения всех параметров, включая содержание масел и воды с учетом назначения и типа газа, конструкции системы приготовления газа и, если нужно, требования действующей фармакопейной статье или требованиям к качеству продукции.
- Газы, используемые в асептическом производстве, должны пройти через стери- лизующие фильтры (с номинальным размером пор не более 0,22 мкм) в точке потребле- ния. Если фильтр используется для производства серии (например, для фильтрования газа в процессе асептического наполнения) или как вентиляционный фильтр в резервуаре, то следует проверять его целостность и результаты рассматривать при сертифика- ции/выпуске серии. Следует стерилизовать все трубы после финишного стерилизующего фильтра. Если газы используются в процессе производства, то следует периодически про- верять их микробиологическую чистоту в точке потребления.
- Следует предусмотреть защиту от обратного потока при выключении систем вакуумирования или сжатого воздуха, если существует риск для продукта.
Системы подогрева, охлаждения и гидравлические системы
6.19.Основные части гидравлических систем или систем подогрева и охлаждения должны, по возможности, находиться за пределами помещений наполнения. Следует при- нять необходимые меры по недопущению проливов и/или перекрестных загрязнений, свя- занных с жидкостями, используемыми в этих системах. 6.20.Должно быть обеспечено обнаружение любых представляющих риск утечек в этих системах (например, системы контроля утечек).7.Персонал
7.1. Производитель должен иметь необходимый персонал, имеющий требуемую квали- фикацию и опыт работы в производстве и контроле качества стерильных лекарственных средств и в любой специальной технологии, используемой на данном производстве, для обес- печения соответствия правилам GMP для производства стерильных лекарственных средств. 7.2. В чистых помещениях допускается нахождение только минимально необходимо- го персонала. Следует определить предельно допустимое число лиц в чистых помещени- ях, оформить это документально и учитывать при первоначальной аттестации и имитации асептического процесса так, чтобы не нарушить условия стерильности. 7.3. Весь персонал, включая персонал, выполняющий уборку, техническое обслужи- вание, контроль и имеющий доступ в чистое помещение должен проходить регулярное обучение, аттестацию на правильность переодевания и оценку по влпросам, связанным с правильным производством стерильной продукции. В программу обучения должны вхо- дить основы микробиологии и гигиены с акцентом на работу в чистых помещениях, кон- троль загрязнений, асептические процессы и защиту стерильной продукции (для операторов, входящих в зоны В и/или имеющих доступ в зоны А) и возможным последствиям для безопасности пациента, если продукт окажется нестерильным. Обучение должно исходить из критичности выполняемых операций и зон, в которых персонал работает. 7.4. Персонал, входящий в чистые помещения с зонами A и B должен пройти обуче- ние порядку переодевания и поведению с учетом требований асептики. Следует проверять правильность переодевания по требованиям асептики, подтверждать это (аттестовать пер- сонал) и проводить периодическую проверку не реже одного раза в год с визуальным и микробиологическим контролем, включая такие дополнительные точки, как предплечья, грудь и капюшон (маска для лица и лоб). Рекомендуемые пределы даны в 9.30. К входу без сопровождения в зоны А и В, в которых выполняются или будут выполняться асепти- ческие операции, допускается только аттестованный персонал, прошедший проверку пра- вильности переодевания и участвовавший в успешной имитации асептического процесса. 7.5. Не допускается вход в зоны В или А неаттестованного персонала при выполне- нии операций. В исключительных случаях и при необходимости производитель может ввести письменные инструкции, устанавливающие порядок входа неаттестованного пер- сонала в зоны А и В. Аттестованное лицо от производителя должно наблюдать за неатте- стованным персоналом при его действиях и должно оценивать влияние этих действий на чистоту зоны. Доступ этих лиц должен быть оценен и оформлен документально согласно системе качества предприятия. 7.6.Следует предусмотреть систему дисквалификации персонала с лишением права работы или входа в чистые помещения без сопровождения, которая должна включать по- стоянную оценку и/или обнаружение отрицательных тенденций по программе контроля персонала и/или после того, как из-за него имитация асептического процесса оказалась неудачной. В случае дисквалификации оператора он может быть допущен к выполнению асептических операций только после прохождения повторного обучения и повторной ат- тестации. Для операторов, входящих в зоны В или выполняющих действия в зоне А, эта повторная аттестация должна включать проверку на успешное участие в имитации асеп- тического процесса. 7.7. Высокие стандарты по личной гигиене и чистоте играют важную роль в предот- вращении избыточных выделений или повышенного риска внесения микробных загрязне- ний. Персонал, занятый в производстве стерильной продукции, должен быть проинструк- тирован о необходимости докладывать о любых особенностях здоровья или заболеваниях, которые могут привести к выделению необычного количества и видов загрязнений, кото- рые могут повлиять на допуск в чистое помещение. Следует возложить на компетентных лиц и оформить в инструкциях требования к состоянию здоровья персонала и принимае- мые к нему действия, если он может внести нежелательные опасные микроорганизмы. 7.8. Не допускается к входу в зоны производства стерильной продукции персонал, который был занят в операциях с материалами тканей человека или животных или куль- турами микроорганизмов, иными, чем используются в текущем технологическом процес- се, или выполнял другие действия, которые могут оказать отрицательное влияние на качество (например, привести к микробному загрязнению), если не выполнены и докумен- тально не оформлены ясные и эффективные порядок обработки и правила входа. 7.9. Не допускается вносить в чистые зоны наручные часы, косметические средства, ювелирные изделия и другие личные предметы, такие как мобильные телефоны, а также предметы, не являющиеся необходимыми. В чистых помещениях могут использоваться электронные устройства (например, мобильные телефоны и планшеты), которые постав- ляются исключительно для чистых помещений и имеют конструкцию, предусматриваю- щую возможность очистки и дезинфекции для данных зон. Использование и дезинфекция таких устройств должно быть учтены в СКЗ. 7.10. Переодевание и мытье рук при входе в чистые помещения должны выполняться в соответствии с письменными инструкциями, предназначенными для сведения к мини- муму загрязнения одежды для чистых зон или переноса загрязнений в чистые зоны. 7.11. Вид одежды и ее качество должны соответствовать процессу и типу рабочей зо- ны. Ее следует носить так, чтобы обеспечить защиту продукции от загрязнений. Если одежда должна защищать оператора от продукта, то при этом не должна снижаться защи- та продукта от загрязнений. Следует визуально проверять одежду на чистоту и целост- ность непосредственно перед ее одеванием и после одевания. Целостность одежды следу- ет также проверять после выхода. Для прошедшей стерилизацию одежды и средств защи- ты для глаз следует с особым вниманием убедиться в том, что время стерилизации выдер- жано и целостность их упаковки проверена визуально до использования. Одежду много- кратного использования (включая защиту для глаз) следует заменять при обнаружении повреждений или с определенной периодичностью, установленной при аттестации. В про- грамму аттестации одежды должны входить все необходимые требования по контролю, включая повреждения одежды, которые не могут быть обнаружены при визуальном осмотре. 7.12.Одеждадолжнаограничиватьвыделенияпривыполнении операций. 7.13. К одежде, предназначенной для зон различных классов чистоты, предъявляются следующие требования:- Зона В (включая доступ и работу в зоне А): до одевания стерильного комбинезона следует надеть нижнюю одежду (п. 7.14). При одевании стерильной одежды следует ис- пользовать стерильные, неопудренные резиновые или полимерные перчатки. Стерильный головной убор должен полностью закрывать все волосы на голове и лице. Если головной убор не является единым целым с одеждой, то его края должны быть убраны под ворот- ник стерильного костюма. Следует носить стерильную маску и стерильную защиту для глаз (например, защитные очки), чтобы закрыть всю кожу лица и предотвратить распро- странение капель и частиц. Следует носить стерильную обувь (например, бахилы). Ниж- няя часть штанин должна быть заправлена внутрь бахил. Рукава одежды должны быть за- правлены под вторую пару перчаток, надеваемую на первую при переодевании. Защитная одежда не должна выделять волокон или частиц и должна удерживать частицы, отделяю- щиеся от тела. При аттестации одежды следует оценить выделение ею частиц и эффектив- ность по удержанию частиц. Одежда должна быть упакована и уложена так, чтобы оператор мог одеть не касаясь наружных поверхностей одежды и не допустить касание одеждой пола.
- Зона С: Волосы, борода и усы должны быть закрыты. Следует носить костюм (комбинезон или куртка – брюки), плотно облегающий запястья, с воротником-стойкой и соответствующим образом дезинфицированные обувь или бахилы. Одежда и обувь не должны выделять волокон или частиц.
- Зона D: Волосы, борода и усы должны быть закрыты. Следует носить защитный костюм общего назначения, и соответствующим образом дезинфицированную обувь или бахилы. Должны быть приняты меры для предотвращения проникания любого загрязне- ния в чистую зону извне.
- При выполнении операций, чувствительных к риску загрязнений, как определено в СКЗ, в зонах С и D могут потребоваться дополнительные принадлежности одежды, включая перчатки и маску.
8.Производство и специальные технологии
Продукция, подлежащая финишной стерилизации (стерилизации в оконча- тельной герметичной первичной упаковке)
8.1. Подготовка компонентов и материалов должны проводиться, по крайней мере, в зоне D, чтобы обеспечить достаточно низкий уровень загрязнения микроорганизмами, эн- дотоксинами/пирогенами и частицами перед стерилизацией. При повышенном или не- обычном риске загрязнения микроорганизмами (например, когда продукт является хоро- шей питательной средой для микроорганизмов или он должен храниться в течение дли- тельного времени до стерилизации, или процесс проводят, в основном, в незакрытом обо- рудовании) приготовление продукта следует выполнять в зоне С. 8.2. Первичные упаковочные материалы и компоненты должны быть очищены (обра- ботаны) аттестованными методами, чтобы снизить до приемлемого уровня загрязнения частицами, эндотоксинами/пирогенами и микроорганизмами. 8.3. Наполнение продуктами, подлежащими финишной стерилизации, должно прово- диться, по крайней мере, в зоне С. 8.4. Если в СКЗ указано, что продукт подвергается необычному (повышенному) рис- ку загрязнения от окружающей среды, например, если операции наполнения проходят медленно или упаковки имеют широкое горло, или их необходимо держать открытыми более нескольких секунд до герметизации, наполнение должно проводиться в зоне А, находящейся, по крайней мере, в зоне С. 8.5. Растворы, которые готовят в балк-форме, подлежат стерилизующей фильтрации, где это возможно, для снижения бионагрузки и концентрации частиц до наполнения в окончательную первичную упаковку. Следует определить максимально допустимое время между приготовлением и наполнением. 8.6.Примерыопераций,выполняемыхвразличныхзонах,приведенывтаблице3.Таблица 3 Примеры операций и зон для продукции, подлежащей финишной стерилизации
|
Зона A |
Наполнение продуктом, когда его нельзя подвергать риску загрязнения |
|
Зона C |
Приготовление растворов, когда их нельзя подвергать риску загрязнения. Наполнение продуктом |
|
Зона D |
Приготовление растворов и компонентов для последующего наполнения |
Асептическое производство
8.7. Асептический процесс должен быть ясно определен. Следует четко установить, оценить и контролировать риски, связанные с асептическим процессом и другими услови- ями. В СКЗ производства должны быть четко указаны критерии приемлемости (допусти- мые пределы) для этих факторов, требования к контролю и рассмотрению его эффектив- ности. Следует дать описание и выполнять методы контроля этих рисков. 8.8. Следует принять меры по сведению к минимуму загрязнений микроорганизмами, эндотоксинами/пирогенами и частицами (как часть СКЗ) при подготовке окружающей среды асептического производства, выполнении всех технологических стадий, включая стадии до и после стерилизующей фильтрации нерасфасованного продукта (балк- продукта) вплоть до герметизации продукта в его окончательной упаковке. Следует све- сти к минимуму нахождение в чистых помещениях материалов, способных выделять ча- стицы и волокна. 8.9. Где это возможно, следует рассматривать использование барьерных систем с ограниченным доступом (RABS), изоляторов или других систем с целью уменьшения вме- шательств в зону А и сведения к минимуму риска загрязнения. Могут так же рассматри- ваться роботизированные или автоматизированные процессы для исключения прямого кри- тического вмешательства человека (например, за счет использования сухожарового тунне- ля, автоматической загрузки лиофилизатора, стерилизации на месте). 8.10. Примеры операций, которые требуют различных зон окружающей среды, при- ведены в таблице 4.Таблица 4 Примеры операций и зоны, в которых они должны выполняться
|
Зона A |
|
|
Зона B |
|
|
Зона C |
- Приготовление растворов для последующей фильтрации, включая отбор проб и их отправку. |
|
Зона D |
ности. |
- Все оборудование, вступающее в контакт с продуктом или компонентами, должно быть стерильным перед использованием.
- Все исходные или промежуточные материалы должны быть стерильными и до- бавлены в асептических условиях.
- Растворы и промежуточные продукты в балк-форме (нерасфасованные) подлежат стерилизации.
При применении изоляторов следует выполнить требования к окружающей среде по п. 4.20. Следует оценивать проверять эффективность асептических соединений. Требова- ния к устройствам, предусматривающим сборку без нарушения стерильности, приведены в 8.129 и 8.130,
8.15. Следует свести к минимуму операции, выполняемые в асептических условиях (включая сборку без нарушения стерильности) за счет инженерных конструкторских ре- шений, таких как предварительная сборка и стерилизация оборудования. Везде, где это возможно, следует выполнять предварительную сборку и стерилизацию на месте трубо- проводов и оборудования, контактирующих с продуктом.
8.16. Должен быть утвержден перечень допустимых аттестованных вмешательств в текущей работе и при выполнении корректирующих действий, которые могут иметь место в процессе производства (п. 9.34). Порядок вмешательства должен быть тщательно разра- ботан, чтобы свести к минимуму риск загрязнения окружающей среды, процесса и про- дукта. При разработке этого порядка следует учитывать влияние любого воздействия на потоки воздуха и критические поверхности и продукты. Везде, где это возможно, следует применять инженерные решения для сведения к минимуму вмешательства оператора в таких случаях. Следует всегда контролировать выполнение асептических требований, включая применение стерильных инструментов для выполнения работы. Виды текущих и корректирующих вмешательств и порядок их выполнения должны содержаться в ин- струкциях, которые подлежат первоначальной оценке путем анализа рисков или имитации асептического процесса и периодические пересматриваться. Неаттестованные вмешатель- ства допускаются лишь в исключительных случаях с учетом фактора риска от этих вме- шательств и при согласовании службой качества. Следует детально оценить риск от вы- полненных вмешательств с документальным оформлением и полным расследованием в рамках системы качества предприятия. Служба качества должна тщательно анализировать любые неаттеставанные вмешательства и учесть при принятии решения о выпуске серии. 8.17. В протоколе серии следует регистрировать вмешательства и перерывы в работе. Следует документально оформлять в протоколе серии каждый случай остановки линии или вмешательства с указанием времени и продолжительности события и оператора (п. 9.34). 8.18. Продолжительность каждого этапа подготовки и выполнения процесса должна быть сведена к минимуму и ограничена аттестованным предельно допустимым временем, включая:- Время между очисткой, сушкой и стерилизацией оборудования, компонентов и первичной упаковки;
- Время нахождения стерильных оборудования, компонентов и первичной упаковки до использования и в процессе наполнения/сборки.
- Время нахождения среды в обработанном состоянии (внутри RABS или изолято- ра) до использования.
- Время между началом приготовления продукта и его стерилизацией или фильтра- цией через удерживающий микроорганизмы фильтр (если требуется) до окончания асеп- тического процесса. Следует установить предельно допустимое время для каждого про- дукта, которые включает его приготовление и предусмотренный метод хранения.
- Время нахождения стерильного продукта до наполнения.
- Времяасептического приготовления.
- Времянаполнения.
Завершающие операции в производстве стерильной продукции
8.20. Открытые контейнеры для первичной упаковки должны находиться в зоне А при окружающей среде, соответствующей применяемой технологии (4.20). Указания для частично укупоренных флаконов или предварительно наполненных шприцев даны в п. 8.126. 8.21. Контейнеры окончательной упаковки должны быть укупорены аттестованными методами. 8.22. Если окончательные упаковки (контейнеры) герметизируются методом запайки, например, пакеты, полученные методами «Выдувание-наполнение-герметизация (Blow- Fill-Seal – BFS)», «Формирование-наполнение-герметизация (Form-Fill-Seal – FFS)» и па- рентеральные препараты в малом и большом объемах (Small and Large Volume Parenteral - SVP & LVP), в стеклянных или пластиковых ампулах, то следует оценить, определить и контролировать в ходе процесса критические параметры и переменные, которые влияют на целостность герметизации. Стеклянные ампулы, упаковки BFS и контейнеры малого объема (≤ 100 мл), герметизируемые запайкой, подлежат 100%-му контролю целостности аттестованными методами. Для контейнеров большого объема (> 100 мл), герметизируе- мых запайкой, может контролироваться меньший объем образцов при условии научного обоснования и наличии данных, показывающих постоянство действующего процесса и высокий уровень соответствия процесса требованиям. Следует заметить, что визуальный контроль не рассматривается как приемлемый контроля целостности. 8.23. Образцы продукции, не относящиеся к получаемой методом запайки, следует отбирать и проверять аттестованными методами. Периодичность контроля должна быть основана на знании используемых систем укупорки и практическом опыте. Следует ис- пользовать научно обоснованный план отбора проб. Количество проб должно быть осно- вано на данных поставщика, спецификаций на компоненты и знании процесса. 8.24. Первичные упаковки, герметизированные под вакуумом (вакуумные упаковки), должны проверяться на сохранение вакуума по истечении заранее установленного перио- да времени до сертификации/выпуска и в течение срока годности. 8.25. Аттестация целостности укупорки контейнера должна учитывать требования транспортирования или отгрузки, которые могут оказать отрицательное влияние на це- лостность контейнера (например, снижение давления или экстремальные температуры). 8.26. Если оборудование для обжима колпачков флаконов может выделять нежизне- способные частицы в больших количествах, то следует принять меры по предотвращению загрязнения частицами, такие как размещение оборудования в физически отделенных зо- нах с необходимой вытяжкой воздуха. 8.27. Установка колпачков для продуктов после асептического наполнения может выполняться как асептический процесс с использованием стерильных колпачков или как чистый процесс за пределами зоны асептического производства. В последнем случае фла- коны должны быть защищены зоной А до границы асептической зоны и далее укупорен- ные флаконы должны быть защищены воздухом, соответствующим зоне А до обжима колпачков. Зону с воздухом, соответствующим зоне А, должна окружать, как минимум, зона D. Ручной обжим колпачков следует выполнять в зоне А, либо в изоляторе соответ- ствующей конструкции, либо в зоне А в окружении зоны В. 8.28. Если установка колпачков на стерильный продукт после наполнения в асепти- ческих условиях выполняется как чистый процесс в зоне А, то флаконы без пробки или со смещенной пробкой следует удалять до установки колпачка. Следует предусмотреть атте- стованные автоматические методы определения высоты установки пробок. 8.29. Если для обжима требуется вмешательство человека, следует предусмотреть меры по предотвращению прямого контакта с флаконами и сведению к минимуму воз- можности загрязнения. Эффективными средствами для обеспечения выполнения этих условий является применение систем с ограниченным доступом (RABS) и изоляторов. 8.30. Все первичные упаковки с парентеральными продуктами следует проверять ин- дивидуально на наличие посторонних включений или других дефектов. Классификация дефектов и оценка их критичности проводятся на этапе аттестации и основываются на анализе рисков и знаниях о предшествующей работе. Следует обратить внимание, как ми- нимум, на такие факторы как возможное влияние на пациента и способ введения. Следует систематизировать различные виды дефектов и анализировать серию продукции.Нужно проводить расследования для серий с необычным уровнем дефектов по срав- нению с текущим числом дефектов (по данным текущей работы и анализа тенденций). Следует вести каталог всех обнаруженных дефектов, который должен включать все из- вестные виды дефектов. Этот каталог следует использовать в качестве учебного пособия для производственного персонала и работников подразделения обеспечения качества. Критические дефекты не должны обнаруживаться при любом последующем отборе проб в признанных годными упаковках (единицах продукции). При обнаружении любого крити- ческого дефекта следует проводить расследование, поскольку это указывает на возможное упущение при первичном контроле.
8.31.Визуальный контроль следует проводить при установленных уровнях освещен- ности и фоне рабочего поля. Темп (скорость) контроля следует контролировать и аттесто- вать. Следует регулярно (не реже одного раза в год) проводить аттестацию операторов, выполняющих визуальный контроль (если операторы носят контактные линзы, то атте- стация проводится в них).При аттестации следует использовать контрольные образцы из архива производите- ля и учитывать ситуации наихудшего случая, например, время контроля, скорость линии (если продукт подается оператору конвейером), размер упаковки, фактор усталости и учитывать результаты проверки зрения. Следует устранить отвлекающие факторы и органи- зовать достаточно частые перерывы работы требуемой длительности.
8.32. Если используются автоматизированные методы контроля, то при аттестации процесса следует показать, что такой метод обнаруживает известные дефекты (которые могут оказать влияние на качество или безопасность) с чувствительностью равной или лучшей, чем при визуальном контроле. Работа оборудования должна быть проверена в начале с использованием представительных дефектов и регулярно проверяться в ходе производства серии. 8.33. Результаты контроля должны быть оформлены документально и отражать виды и тенденцию изменения числа дефектов. Следует оценивать тенденции появления различных дефектов с использованием статистических принципов. При анализе отрицательных тенден- ций следует оценивать влияние дефектов на продукцию на рынке.Стерилизация
8.34. Везде, где это возможно, следует предусматривать финишную стерилизацию готовой продукции с использованием аттестованного и контролируемого процесса стери- лизации, поскольку это обеспечивает более высокую гарантию стерильности, чем аттесто- ванный и контролируемый процесс стерилизующей фильтрации и/или асептический про- цесс. Если финишная стерилизация продукции не допустима, то следует рассмотреть воз- можность тепловой обработки после асептического процесса для повышения гарантии стерильности. 8.35. Выбор оборудования, его конструкция и расположение оборудования и цикл (программа) стерилизации должны быть основаны на научных принципах и данных, де- монстрирующих повторяемость и надежность процесса стерилизации. Все параметры должны быть заданы, а критические параметры следует контролировать и регистрировать. 8.36. Все процессы стерилизации должны быть аттестованы. При аттестации следует учесть состав продукта, условия хранения и максимальное время между началом приго- товления продукта или материала, подлежащего стерилизации, и его стерилизацией.До применения любого процесса стерилизации следует показать его пригодность для данной продукции и оборудования и эффективность в постоянном достижении требуемых условий стерилизации во всех частях каждого типа загрузки (с помощью физических из- мерений и, если возможно, биологических индикаторов). Для обеспечения эффективной стерилизации следует предусмотреть необходимую обработку всех материалов и оборудова- ния, что следует учесть при разработке процесса.
8.37. Требуется особое внимание, если применяемый метод стерилизации не описан в действующей Фармакопее или используется для продукта, не являющегося простым вод- ным раствором. Предпочтительным является метод тепловой стерилизации. 8.38.Следует применять аттестованные схемы загрузки для всех процессов стерили- зации. Схемы загрузки подлежат периодической повторной аттестации. Схемы максимальной и минимальной загрузок следует рассматривать как часть общей стратегии аттестации загрузки. 8.39. Следует рассматривать и подтверждать соответствие процесса стерилизации установленным требованиям через определенные интервалы времени с учетом фактора риска. Повторную аттестацию процессов тепловой стерилизации следует проводить не реже одного раза в год для схемы загрузки, представляющей наихудший случай. Для дру- гих схем загрузки периодичность повторной аттестации должна быть указана в стратегии контроля загрязнений. 8.40.Следует разработать и выполнять требования ко всем эксплуатационным пара- метрам для всех процессов стерилизации с указанием, например, требований к физиче- ским параметрам, схемам загрузки и др. 8.41.Следует предусмотреть порядок обнаружения циклов стерилизации, которые не соответствуют аттестованным параметрам. Любой неудачный цикл стерилизации или цикл с отклонениями от аттестованного процесса (например, более короткие или более длительные периоды времени на нагрев) должен быть расследован. 8.42.Биологические индикаторы (биоиндикаторы), помещенные в определенные точки, являются дополнительными средствами при аттестации процесса стерилизации. Биоиндикаторы должны храниться и использоваться в соответствии с инструкциями изго- товителя. Следует подтверждать качество новой серии биоиндикаторов до начала их ис- пользования путем счета жизнеспособных спор и их идентификации. Если биоиндикаторы используются для аттестации и/или контроля процесса стерилизации (например, оксидом этилена), то следует проводить положительный контроль для каждого цикла стерилиза- ции. При использовании биоиндикаторов следует предусмотреть меры предосторожности против переноса микробных загрязнений в производство или на другие процессы кон- троля. Результаты контроля с помощью биоиндикаторов не заменяют оценки других кри- тических параметров и факторов, определяющих процесс. 8.43.Надежность биоиндикаторов играет важную роль. Следует выполнять аттеста- цию поставщиков и контролировать условия транспортирования и хранения, чтобы не до- пустить отклонений от требований к качеству биоиндикаторов. Перед использованием но- вой серии (партии) биоиндикаторов следует проверить число, чистоту и идентичность ин- дикаторных микроорганизмов. Для других критических параметров, например, величины D, величины Z может, как правило, использоваться сертификат серии аттестованного по- ставщика. 8.44.Следует четко определить меры, обеспечивающие разделение продуктов, обо- рудования и компонентов, прошедших и не прошедших стерилизацию. На каждой кор- зине, лотке для перемещения продукта, другие частях оборудования и/или компонентах должна быть четкая этикетка (или электронный код) с наименованием продукта, номером серии и указанием, прошел он стерилизацию или нет. Для обозначения, прошла ли серия (или подсерия) процесс стерилизации или нет, можно использовать такие индикаторы, как автоклавная лента или радиоактивные индикаторы, где это требуется. Эти индикаторы показывают лишь то, что процесс стерилизации прошел, но не подтверждают стерильность продукта или достижения требуемого уровня стерильности. 8.45.Следует вести протоколы для каждого цикла стерилизации. Каждый цикл должен иметь свой собственный код. Соответствие циклов требованиям должно рассматриваться и утверждаться как часть процесса выпуска серии (сертификации). 8.46.Где требуется, стерилизация материалов, оборудования и компонентов должна выполняться аттестованным методом, соответствующим данному материалу. Для преду- преждения загрязнений следует предусмотреть защиту после стерилизации. Если предме- ты, прошедшие стерилизацию, не используются сразу после стерилизации, то их следует хранить в герметичной упаковке с установлением максимального времени хранения. До- пускается, при обосновании, не хранить компоненты в чистом помещении при наличии многослойной стерильной упаковки материалов и обоснования, если целостность и схема стерильной упаковки позволяют ее легко дезинфицировать при передаче в зону А (напри- мер, в случае многослойной стерильной упаковки, каждый слой которой может удаляться при перемещении из более низкой зоны в более высокую). Если защита обеспечивается помещением в герметичную упаковку, то это должно быть выполнено до стерилизации. 8.47.Если материалы, оборудование, компоненты и вспомогательные вещества под- лежат стерилизации в герметичной упаковке и потом передаются в зону А, то это следует выполнять аттестованными методами (например, через воздушные шлюзы или передаточ- ные камеры) с проведением дезинфекции внешних поверхностей герметичной упаковки. Следует учесть применение технологии с использованием портов (средств) быстрой пере- дачи. Следует показать эффективность этих мер по предупреждению риска потенциально- го загрязнения зон А и В, а также эффективность метода дезинфекции по снижению за- грязнения упаковки до приемлемого уровня для передачи предметов в зоны В и А. 8.48.Если материалы, оборудование, компоненты и вспомогательные предметы под- лежат стерилизации в герметичной упаковке или в контейнерах, то следует аттестовать целостность стерильного защитного барьера по сведению к минимуму риска загрязнения частицами, микроорганизмами, эндотоксинами/пирогенами или химическими веществами и на совместимость с применяемым методом стерилизации. Процесс стерилизации упа- ковки должен быть аттестован. Аттестация должна учесть целостность стерильного за- щитного барьера, максимальный период времени до стерилизации и максимальный срок хранения стерильных предметов. Перед использованием следует проверить целостность системы стерильного защитного барьера. 8.49.Для материалов, оборудования, компонентов и вспомогательных предметов, ко- торые не вступают в прямой или непрямой контакт с частями продукта, но которые тре- буются в асептическом процессе и которые не допускают стерилизации, следует приме- нять эффективные и аттестованные методы дезинфекции и передачи. После дезинфекции должна быть предусмотрена защита этих предметов от загрязнений. Эти предметы, а так- же другие источники возможного загрязнения, должны быть включены в программу кон- троля окружающей среды.Тепловая стерилизация
8.50.Каждый цикл тепловой стерилизации должен быть записан в электроном виде или на твердом носителе с использованием оборудования, имеющего необходимую точ- ность. Система должна быть обеспечена средствами контроля, которые имеют защиту и/или избыточность, чтобы обнаруживать цикл, несоответствующий требованиям к пара- метрам аттестованного цикла и исключать или браковать этот цикл (например, используя дублирование датчиков, соединенных с независимыми системами мониторинга). 8.51.Расположение датчиков температуры, используемых для контроля и/или записи, должно быть определено во время аттестации с учетом конструкции системы для пра- вильной регистрации и представления текущих условий цикла. При аттестации следует показать правильность выбора точек расположения датчиков контроля и записи и прове- рить их работу с помощью независимых датчиков, располагаемых в тех же точках. 8.52.Вся загрузка должна достигать заданной температуры до начала измерений для периода стерилизации. Для циклов стерилизации, использующих контрольные (эталон- ные) датчики внутри загрузки, следует до начала цикла удостовериться в том, что датчик контролирует температуру в загрузке в заданных пределах. 8.53.После завершения высокотемпературной фазы цикла тепловой стерилизации следует принять меры предосторожности от загрязнения стерильной загрузки при охла- ждении. Любая охлаждающая жидкость или газ, которые вступают в контакт с продуктом или стерилизуемым материалом, должны быть стерилизованы. 8.54.Если разрешен выпуск продукции по параметрам, то должна действовать четкая система аттестации жизненного цикла и текущего контроля производственного процесса. Эту систему следует периодически пересматривать. Руководство по выпуску продукции по параметрам дано в приложении 17.Стерилизация влажным теплом
8.55.Стерилизация влажным теплом может выполняться с помощью пара (прямой и непрямой контакт), а также другими способами, например, системами перегретой воды (каскадный или иммерсионный циклы) для контейнеров, которые могут быть повреждены при использовании иных методов (например, контейнеры по технологии «выдувание- наполнение-герметизация» - BFS, пластиковые мешки). 8.56.Стерилизуемые предметы, не являющиеся продуктами в герметичных контей- нерах, должны быть сухими, упакованными в защитную барьерную систему, которая до- пускает удаление воздуха и проникание пара и предупреждает загрязнение после стерили- зации. Все предметы загрузки должны быть сухими до удаления из стерилизатора. Су- хость загрузки должна быть подтверждена визуальными осмотром, который является ча- стью процесса приемки. 8.57.При стерилизации пористой загрузки (твердые предметы) следует контролиро- вать и регистрировать время, температуру и давление. После удаления из автоклава каж- дый предмет следует проверить на отсутствие повреждений, герметичность, целостность упаковки и удаление влаги. Герметичность и целостность упаковки следует также прове- рять непосредственно перед использованием. Не соответствующие этим условиям пред- меты следует удалить из производственной зоны и провести расследование причин. 8.58.В автоклавах, которые предусматривают предварительное вакуумирование в цикле стерилизации, следует регистрировать температуру в зоне стока из камеры в тече- ние всего периода стерилизации. Могут также использоваться датчики внутри загрузки, если требуется, но система контроля должна соответствовать условиям, поученным при аттестации загрузки. Для систем стерилизации на месте следует регистрировать темпера- туру в точках стока в течение всего времени стерилизации. 8.59.При аттестации циклов стерилизации пористой загрузки следует определить время до достижения равновесного состояния, время выдержки, установить связь между давлением и температурой и минимальные/максимальные значения температуры при сте- рилизации. При аттестации циклов стерилизации жидкостей следует определять темпера- туру, время и/или величину F0. Критические параметры процесса должны находиться в заданных пределах (включая допустимые отклонения), что должно быть подтверждено при аттестации процесса стерилизации и текущем контроле. 8.60.Если в цикл стерилизации входит этап вакуумирования или система после сте- рилизации возвращается к давлению, равному или меньшему, чем давление окружающей среды, то следует периодически (как правило, один раз в неделю) проводить проверки ка- меры на герметичность. 8.61.Если цикл стерилизации включает в себя удаление воздуха (например, для по- ристой загрузки, камер лиофилизаторов), то должна быть обеспечена гарантия удаления воздуха до и в процессе стерилизации. Для автоклавов следует выполнять тест на удале- ние воздуха (как правило, ежедневно) или применять детекторы наличия воздуха. Загруз- ки для стерилизации должны быть сформированы так, чтобы обеспечить эффективное удаление воздуха и иметь свободный сток для предотвращения накопления конденсата. 8.62.Для предотвращения деформации и повреждения гибких упаковок, таких как при технологии «выдувание – наполнение – герметизация» и «формирование – наполне- ние – герметизация» и которые подлежат финишной стерилизации, следует предусматри- вать необходимые циклы стерилизации и методы контроля (например, установку требуе- мого давления, скоростей нагрева и охлаждения и схему загрузки). 8.63.Конструкция системы, для которой применяется стерилизация на месте – SIP (например, жестко установленных трубопроводов, сосудов и камер лиофилизаторов), должна обеспечивать требуемую обработку всех частей системы, что должно быть под- тверждено при аттестации. Следует предусмотреть контроль температуры, давления и времени в критических точках такой системы при ее работе для гарантии того, что стери- лизация выполняется эффективно и воспроизводимым образом. При первоначальной и повторной аттестации следует показать, что эти критические точки являются представи- тельными и соответствуют местам с самым медленным прогревом. После завершения сте- рилизации системы с помощью SIP она должна оставаться целостной (герметичной) и, где требуется, находиться под положительным давлением до использования или иметь стери- лизующий вентиляционный фильтр. 8.64.При использовании перегретой воды как средства передачи тепла при стерили- зации жидкостей, нагретая вода должна непрерывно поступать ко всем точкам контакта. При первоначальной аттестации следует построить карту температур для всей загрузки. При текущей работе следует проверять, что форсунки, через которые подается вода, не засорены и сток не засорен. 8.65.Аттестация процесса стерилизации жидкости с помощью перегретой воды должна включать построение карты температур для всей загрузки и анализ проникания тепла и воспроизводимости процесса. Все части загрузки должны прогреваться равномер- но и достигать заданной температуры в заданное время. Датчики текущего контроля должны находиться в местах наихудшего случая, установленных при аттестации.Сухожаровая стерилизация
8.66.Для стерилизации продукта или предмета при сухожаровой стерилизации тре- буется высокая температура воздуха или газа. Сухожаровая стерилизация предназначена для термического удаления загрязнений, устойчивых к воздействию тепла, таких как эн- дотоксины/пирогены, и часто используется при подготовке компонентов для асептическо- го наполнения. Сочетание времени и температуры, при которых выдерживаются продукт, компоненты или оборудование, должно обеспечивать адекватный и воспроизводимый уровень летальности и/или инактивацию/удаление эндотоксинов/пирогенов в текущей ра- боте в установленных пределах. Процесс может выполняться в шкафу или в туннеле (не- прерывный процесс), например, для стерилизации и депирогенизации стеклянных контей- неров. 8.67.Конструкция туннелей сухожаровой стерилизации/депирогенизации должна обеспечивать защиту воздушным потоком целостности и условий зоны стерилизации А за счет поддержания требуемого давления и движения потока воздуха во всем туннеле. Сле- дует оценить значения перепадов давления воздуха. Следует оценить влияние изменений потоков воздуха, чтобы обеспечить поддержание требуемых условий нагрева. Весь воз- дух, подаваемый в туннель, должен проходить через НЕРА фильтры. Следует проверять целостность воздушных фильтров не реже, чем один раз в два года. Любые части туннеля, вступающие в контакт со стерилизуемыми предметами, должны быть стерилизованы или дезинфицированы требуемым образом. К критическим параметрам процесса, которые следует учесть при аттестации и/или текущей работе, относятся (перечень не исчерпывающий):- Скорость движения конвейера или время нахождения в зоне стерилизации;
- Максимальное и минимальное значения температуры;
- Проникание тепла в материал/предмет;
- Однородность распределения тепла;
- Потоки воздуха, определенные по перепадам давления воздуха, в зависимости от анализа распределения и проникания тепла.
- Температура;
- Время выдержки;
- Давление в камере (поддержание положительного давления);
- Скорость воздуха;
- Качество воздуха внутри шкафа;
- Проникание тепла в материал/предмет (места с медленным нагревом);
- Распределение/однородность тепла;
- Схема загрузки и конфигурация предметов, подлежащих стерилиза- ции/депирогенизации, включая минимальную и максимальную загрузки.
Радиационная стерилизация
8.71.Радиационную стерилизацию используют главным образом для стерилизации термочувствительных материалов и продуктов. Ультрафиолетовое облучение не является приемлемым методом стерилизации. Руководство по ионизирующей радиационной стери- лизации приведено в приложении 12. 8.72.Методы аттестации (испытаний) должны гарантировать учет возможных изме- нений плотности продукта и упаковки.Стерилизация этиленоксидом
8.73.Этот метод может использоваться только в том случае, если нельзя применять другие методы стерилизации. При проведении аттестации (испытаний) следует показать, что процесс стерилизации этиленоксидом не оказывает вредного влияния на продукцию, а заданные условия и длительность дегазации позволяют снизить остаточную концентрацию газа и продуктов реакции до допустимых уровней, определенных для данного вида продук- ции или материала. 8.74.Существенное значение имеет непосредственный контакт между газом и мик- роорганизмами. Следует принять меры предосторожности от включения микроорганизмов в материал (например, в кристаллы или лиофилизированный белок). На процесс стерили- зации могут оказать существенное влияние вид, пористость и количество упаковочного материала. 8.75.Температуру и влажность загрузки перед обработкой газом следует привести в соответствие с требованиями процесса. В случае использования пара для кондиционирова- ния загрузки он должен иметь необходимое качество. Требуемое для этого время должно быть определено с учетом сведения к минимуму всего времени до стерилизации. 8.76.Каждый цикл стерилизации должен контролироваться с использованием соот- ветствующих биологических индикаторов, распределенных в достаточном количестве по всему объему загрузки в определенных точках, для которых при аттестации показано, что они являются точками «наихудшего случая». 8.77.К критическим параметрам процесса, которые должны быть учтены при атте- стации и/или в текущей работе, относятся (перечень не исчерпывающий):- Концентрация этиленоксида (газа);
- Давление;
- Использованное количество этиленоксида;
- Относительная влажность;
- Температура;
- Время выдержки (стерилизации).
8.78 После стерилизации загрузку следует подвергнуть аэрации, чтобы обеспечить удаление этиленоксида (газа) и/или продуктов реакции из упакованного продукта до зара- нее установленного уровня.
Стерилизующая фильтрация продуктов, которые не могут быть стерилизованы в окончательной упаковке
8.79.Если продукт не может быть стерилизован в его окончательной упаковке, то растворы или жидкости следует стерилизовать путем фильтрации через стерилизующие фильтры (с номинальным размером пор 0,22 мкм, которые аттестованы на предмет получения стерильного фильтрата) с последующим асептическим наполнением в предвари- тельно стерилизованный контейнер. Выбор фильтра должен гарантировать, что он совме- стим с продуктом, как указано в лицензии на реализацию (см п. 8.135) 8.80.В различных точках технологического процесса могут использоваться пред- фильтры, снижающие бионагрузку и/или стерилизующие фильтры, чтобы обеспечить низкую и контролируемую бионагрузку в жидкости до поступления на финишный стери- лизующий фильтр. В связи с тем, что процесс стерилизующей фильтрации создает допол- нительный риск по сравнению с другими методами стерилизации, рекомендуется преду- сматривать дополнительную фильтрацию через стерилизующий фильтр как можно ближе к точке наполнения. Это должно рассматриваться как часть СКЗ. 8.81.Выбор элементов для систем фильтрации, их соединение друг с другом и ком- поновка в системе фильтрации, включая предфильтры, должны основываться на критиче- ских свойствах продукта для качества, обоснованы и документально оформлены. Выделе- ние волокон и частиц в системе фильтрации должно быть сведено к минимуму. Система не должна приводить к увеличению загрязнений до неприемлемого уровня или способ- ствовать этому или влиять на характеристики, которые могут изменить качество и эффек- тивность продукта. Аналогично этому, характеристики фильтра должны быть совместимы с жидкостью и не оказывать отрицательного влияния на подлежащий фильтрации про- дукт. Следует оценить абсорбцию компонентов продукта и экстракцию/выделение компо- нентов фильтра (см. п. 8.135). 8.82Конструкция системы фильтрации должна:- Обеспечивать работу в пределах аттестованных значений параметров процесса;
- Обеспечивать стерильность фильтрата;
- Сводить к минимуму число асептических соединений между финишным стерили- зующим фильтром и местом окончательного наполнения продуктом;
- Допускать выполнение очистки в требуемом объеме;
- Допускать выполнение стерилизации, включая стерилизацию на месте, в требуе- мом объеме;
- Позволять проведение контроля целостности «на месте» финишных стерилизую- щих фильтров на 0,22 мкм, предпочтительно, как закрытой системы, как до, так и после фильтрации, при необходимости. Следует применять методы контроля целостности «на месте», чтобы исключить отрицательное влияние на качество продукта.
8.85.Ккритическимпараметрампроцессафильтрации,которыедолжныбыть учтены при аттестации относятся (перечень не исчерпывающий):
- Увлажняющая жидкость, используемая для контроля целостности фильтра:
- Должна соответствовать рекомендациям изготовителя фильтра или являться жидкостью подлежащей фильтрации;
- Если система увлажняется или проверяется на целостность «на месте» с по- мощью жидкости, не являющейся продуктом, то следует принять меры по ис- ключению отрицательного влияния на качество продукт
-
- Время нахождения жидкости перед фильтрацией и его влияние на бионагруз- ку;
- Подготовку (conditioning) фильтра, при необходимости;
- Минимальное и общее время нахождение фильтра в контакте с жидкостью;
- Максимально рабочее давление;
- Расход жидкости;
- Максимальный объем при фильтрации;
- Температуру;
- Время, требуемое для фильтрации определения объема раствора и перепад давления на фильтре.
- Глубокие знания процесса стерилизующей фильтрации и его контроль для сведе- ния к минимуму возможности повреждения фильтра;
- Глубокие знания и контроль цепи поставки, включающие:
- Условия проведения стерилизации по контракту;
- Средства транспортирования;
- Упаковку стерилизующего фильтра для предотвращения повреждения филь- тра при транспортировании и хранении;
-
- Специфических характеристик продукции, включая наличие частиц, и суще- ствования риска влияния на показатели целостности фильтра, например, воз- можности изменять результаты контроля целостности и поэтому маскировать обнаружение поврежденного фильтра при контроле целостности после ис- пользования;
- Стадий предварительной фильтрация и процесса до финишного стерилизую- щего фильтра, на которых могут удаляться частицы и которые могут способ- ствовать чистоте продукта, до стерилизующей фильтрации.
8.93.Пробы на микробиологический контрольследует отбирать из нерасфасованного продукта (балк-продукта) и непосредственного перед финишной стерилизующей филь- трацией. При резервировании фильтров пробы следует отбирать до первого фильтра. Си- стема отбора проб должна быть построена так, чтобы не вносить загрязнений.
8.94.Стерилизующие фильтры для жидкостей должны быть удалены после оконча- ния производства одной серии. Не допускается использовать один и тот же фильтр в тече- ние более одного рабочего дня, за исключением случаев, когда такая возможность под- тверждена аттестацией. 8.95. Если возможность выпуска продукции циклами обоснована в СКЗ и аттестова- на, то пользователю фильтров следует:- Оценить b оформить документально риски, связанные с длительностью использо- вания фильтров для стерилизующей фильтрации данной жидкости;
- Выполнить и оформить документально эффективные исследования и аттестацию, подтверждающую, что длительность использования фильтров для данного процесса сте- рилизующей фильтрации и данной жидкости не оказывает отрицательного влияния на финишный стерилизующий фильтр или качество фильтрата;
- Оформить документально аттестованную максимальную длительность использо- вания фильтра и установить контроль, обеспечивающий неиспользование таких фильтров за пределами аттестованной максимальной длительности.
- Установить контроль, обеспечивающий удаление и не допускающий использова- ние фильтров, загрязненных жидкостью или остатками моющих средств, или с наличием других дефектов.
Технология «Формирование-наполнение-герметизация» (Form-Fill-Seal – FFS)
8.96.Условия для оборудования FFS для продукции, подлежащей финишной стери- лизации, должны соответствовать требованиям п.п. 8.3 и 8.4 данного приложения. Усло- вия для оборудования FFS для асептического производства должны соответствовать тре- бованиям к окружающей среде по п. 8.10 данного приложения. 8.97.Загрязнение упаковочных пленок, используемых в процессе FFS, должно быть сведено к минимуму за счет контроля при изготовлении компонентов, их поставке и об- ращении. Ввиду критической роли упаковочных пленок следует ввести порядок, обеспе- чивающий поставку пленок, отвечающих утвержденным спецификациям и требованиям к качеству, включая толщину и прочность пленок, загрязнению микроорганизмами и части- цами, целостности и художественное оформление, если требуется. Следует определить и контролировать в фармацевтической системе качества и СКЗ периодичность отбора проб, уровни загрязнения микроорганизмами и, если требуется, эндотоксинов/пирогенов на упаковочных пленках и связанных с ними компонентах. 8.98.Особое внимание следует уделить пониманию и оценке работы с оборудовани- ем, включая процессы подготовки, наполнения, герметизации и разрезания так, чтобы па- раметры критических процессов были уяснены, аттестованы и подлежали контролю, в т. ч. текущему.8.99.Следует фильтровать любые вступающие в контакт с продуктом газы, напри- мер, используемые для надувания контейнера или в качестве заполнения пространства вблизи продукта, как можно ближе к точке использования. Следует периодически прове- рять качество газов и эффективность фильтрующей системы согласно п.п. 6.18 и 6.19.
8.100.Методы контроля, определенные при аттестации FFS, должны соответствовать СКЗ. Требуется учитывать следующие факторы:- Определение границ критической зоны;
- Контроль окружающей среды, в т. ч. текущий, как оборудования, так и зон, в ко- торых оно находится;
- Требования к одежде персонала;
- Контроль целостности линий наполнения продуктом и фильтрационных систем (если требуется);
- Продолжительность выпуска серии или цикла наполнения;
- Контроль упаковочных пленок, включая любые требования к дезинфекции пле- нок или стерилизации;
- Очистку или стерилизацию оборудования на месте, если требуется;
- Работу с оборудованием, подготовку к работе и установление сигналов тревоги (если требуется).
- Обеспечение одинаковых размеров упаковок и разрезания в соответствии с атте- стованными параметрами;
- Организацию технического обслуживания и контроля аттестованных температур формования (включая предварительный нагрев и охлаждение), значения времени и давле- ния формования, если требуется;
- Организацию технического обслуживания и контроля однородности аттестован- ных температур по контуру герметизации, значений времени и давления герметизации, если требуется;
- Температура окружающей среды и продукта;
- Специфические для серии испытания прочности и однородности герметизации упаковки;
- vi.Обеспечениетребуемыхобъемов,скоростейиоднородностинаполнения;
- Обеспечение нанесения любых дополнительных печатных надписей (кода се- рии), тиснения или снятия тиснения так, чтобы не нарушить целостность упаковки;
- Методы и параметры для контроля целостности наполненных контейнеров (п.8.22).
8.102.В производстве следует применять необходимые методы аттестации, текущего контроля и регистрации критических процессов FFS и работы оборудования.
8.103.Методы обнаружения и устранения дефектов при формовании и герметизации следует отразить в технологических инструкциях. Необходимо регистрировать и рассле- довать случаи отбраковки или нарушения герметизации упаковок. 8.104.Должны быть разработаны инструкции по техническому обслуживанию, осно- ванные на анализе рисков, которые должны включать графики и технического обслужи- вания и проверок критических инструментов на эффективность герметизации упаковок. Любые недостатки, связанные с потенциальной угрозой качеству, нужно регистрировать и расследовать.Выдувание-наполнение-герметизация (Blow-Fill-Seal – BFS)
8.105.Оборудование «выдувание-наполнение-герметизация», используемое для про- изводства продукции, подлежащей финишной стерилизации, должно быть установлено, по крайней мере, в зоне D. Условия в точке наполнения должно соответствовать требова- ниям п.п. 8.3 и 8.4. 8.106.ОборудованиеBFS,используемоевасептическихпроцессах, должно: 8.107.Для оборудования челночного типа, используемого в асептических процессах, за- готовка для контейнера (трубка) открыта в окружающую среду, поэтому экструзия труб- ки, выдувание-формование и герметизация должны выполняться в зоне А в критических зонах. Проект и обслуживание зоны наполнения должны соответствовать требованиям к зоне А по микроорганизмам и общему числу частиц как в оснащенном, так и в эксплуати- руемом состояниях.- Для оборудования роторного типа, используемого в асептическом наполнении, за- готовка для контейнера (трубка) обычно является закрытой по отношению к окружающей среде. Область внутри трубки должна соответствовать требованиям к зоне А по микроор- ганизмам и общему числу частиц как в оснащенном, так и в эксплуатируемом состояниях, что должно обеспечиваться ее проектом и обслуживанием.
- Оборудование должно быть установлено, по крайней мере, в зоне С, одежда должна соответствовать требованиям к зонам А/В. Текущий микробиологический кон- троль операторов в зоне С, одетых в одежду для зон А/В, должен выполняться согласно принципам анализа рисков. Допустимые пределы и периодичность контроля должны учи- тывать активность этих операторов
- При текущем контроле в эксплуатации для оборудования BFS не устанавлива- ются пределы для общего числа частиц, поскольку во время полимерной экструзии и рез- ки выделяются частицы, и размеры зон наполнения в оборудовании BFS ограничены. Од- нако, следует иметь данные, показывающие, что конструкция оборудования обеспечивает выполнение условий зоны А в эксплуатации в критических зонах окружающей среды процесса наполнения.
3.108.Контроль жизнеспособных частиц в процессах BFS следует основывать на анализе рисков и выполнять согласно разделу 9 настоящего приложения. Текущий кон- троль жизнеспособных частиц в эксплуатации должен выполняться течение всего крити- ческого процесса, включая сборку оборудования. Для оборудования BFS роторного типа текущий контроль критической зоны наполнения может оказаться невозможным.
8.109.В программах контроля (текущего контроля) следует учитывать движущиеся части и сложные потоки воздуха, возникающие в ходе процесса BFS, и влияние высокого тепловыделения во время процесса (например, выполнять визуализацию потоков воздуха и/или другой анализ работы оборудования). Программы текущего контроля окружающей среды должны учитывать такие факторы как форму воздушных фильтров, целостность воздушных фильтров, целостность систем охлаждения (см. п. 6.21), конструкцию и атте- стацию оборудования. 8.110.Воздух или другой газ, который вступает в контакт с критическими поверхно- стями контейнера во время экструзии, формовки или герметизации, должен проходить фильтрацию. Качество газа и эффективность фильтрующей системы газа должны перио- дически проверяться согласно п.п. 6.18 и 6.19. 8.111.Следует предотвращать загрязнение полимерного гранулята частицами и мик- роорганизмами за счет проектных решений, контроля и технического обслуживания при хранении, отборе проб и распределения. 8.112.Следует понимать и аттестовать способность экструзионной системы обеспе- чивать требуемую стерильность формованных контейнеров. Следует определить перио- дичность отбора проб, уровни загрязнений микроорганизмами и частицами и, если требу- ется, эндотоксинов/пирогенов полимерного сырья и контролировать их в рамках системы фармацевтического качества и СКЗ. 8.113.Вмешательства, требующие остановки процесса наполнения и/или экструзии, формовки и герметизации и, если требуется, повторной стерилизации оборудования наполнения, следует четко определить и описать в инструкции по наполнению и включить в программу имитации асептического процесса - АРS, если требуется (п.п. 9.34, 9.35 и 9.36). 8.114.Методы контроля, определенные при аттестации BFS, должны соответствовать СКЗ производства. При этом нужно учесть следующее (перечень неисчерпывающий):- Определение границ критической зоны;
- Контроль окружающей среды, в т. ч. текущий, как оборудования, так и зон, в ко- торых оно находится;
- Требования к одежде персонала;
- Контроль целостности линий наполнения продуктом и фильтрационных систем (если требуется);
- Продолжительность выпуска серии или цикла наполнения;
- Контрольполимерного гранулята, включая системураспределения икритические температуры экструзии;
- Очистку или стерилизацию оборудования на месте, если требуется;
- Работу с оборудованием, подготовку к работе и установление сигналов тревоги (если требуется).
- Очистка и стерилизация на месте трубопроводов для продукта и игл наполнения (наконечников);
- Организация технического обслуживания и контроля аттестованных параметров, включая температуру, скорость и регулировку отверстия экструдера для установки тол- щины трубки;
- Организация технического обслуживания и контроля температур формования, включая скорость охлаждения для обеспечения стабильности продукта, при необходимо- сти;
- Приготовление и стерилизация вспомогательных компонентов, добавляемых в формованную единицу, например, колпачков на бутылки;
- Контроль окружающей среды, очистка, стерилизация и текущий контроль в кри- тических зонах экструзии, передачи и наполнения, если требуется;
- Специфический для серии контроль толщины стенок упаковки в критических точках контейнера;
- Обеспечение требуемых объемов, скоростей и однородности наполнения;
- Обеспечение нанесения любых дополнительных печатных надписей (кода се- рии), тиснения или снятия тиснения так, чтобы не нарушить целостность упаковки;
- Методы и параметры для контроля целостности 100% наполненных контейнеров (п. 8.22).
- Установка резцов или перфораторов для удаления остатков пластмассы вокруг наполненных единиц (удаление отливов).
- В асептических процессах добавление компонентов должно выполняться в зоне А для обеспечения стерильности критических поверхностей, используя предварительно сте- рилизованные компоненты.
- Для продукции, подлежащей финишной стерилизации, аттестация процессов фи- нишной стерилизации должна показать стерильность всех критических путей прохожде- ния продуктом между компонентом и формованным контейнером, включая зоны, которые не увлажняются при стерилизации.
- Следует определить и аттестовать методы контроля для обеспечения эффектив- ной герметизации компонентов и формованных контейнеров.
Лиофилизация
8.121.Лиофилизация является критической стадией процесса и все операции, кото- рые могут оказать влияние на стерильность продукции или материалов следует рассмат- ривать как продолжение асептического процесса производства стерильной продукции. Конструкция оборудования для лиофилизации и процессы должны обеспечивать сохране- ние стерильности продукции или материалов при лиофилизации, предотвращение загряз- нения микроорганизмами и частицами между наполнением продукции для лиофилизации и завершением процесса лиофилизации. Все меры по контролю на месте должны быть указаны в CКЗ. 8.122.Процесс стерилизации лиофилизатора и связанного с ним оборудования (например, поддоны, кольца, поддерживающие флаконы) должен быть аттестован и при имитации асептического процесса должно быть определено время между циклом стерили- зации и использованием (п. 9.33). Следует регулярно проводить стерилизацию лиофилиза- тора, исходя из конструкции системы. После выполнения технического обслуживания или очистки следует проводить повторную стерилизацию. После окончания стерилизации лиофилизатор и связанное с ним оборудование должно быть защищено от загрязнений. 8.123.Конструкция лиофилизаторов и зон передачи продукта, разгрузки и выгрузки должна, по возможности, сводить к минимуму вмешательство операторов. Следует опре- делить периодичность стерилизации лиофилизатора исходя из его конструкции и рисков загрязнения системы при использовании. Лиофилизаторы с ручной загрузкой или выгруз- кой без разделения за счет барьерной технологии следует стерилизовать перед каждой за- грузкой. В случаях использования автоматических систем загрузки и выгрузки или защиты с применением закрытых барьерных систем, периодичность стерилизации должна быть обоснована и оформлена документально как часть СКЗ. 8.124.Целостность лиофилизатора следует поддерживать после стерилизации и в процессе лиофилизации. Фильтр, обеспечивающий целостность лиофилизатора, следует стерилизовать перед каждым использованием системы. Результаты контроля целостности фильтра следует включить в документацию на серию/реализацию. 8.125.Следует регулярно проверять поддоны для лиофилизации на предмет сохране- ния формы и отсутствия повреждения. 8.126.Порядок загрузки лиофилизатора (и выгрузки, если лиофилизированный про- дукт не находится в герметичной упаковке и открыт в окружающую среду) должен вклю- чать в себя следующее (перечень неисчерпывающий):- Процесс загрузки должен быть определен и оформлен документально;
- Передача частично закрытых контейнеров в лиофилизатор должна выполняться в зоне А на всем протяжении и порядок передачи должен сводить к минимуму прямые вмешательства оператора. Для обеспечения чистоты системы, используемой для транс- портирования частично закрытых контейнеров, должны использоваться технологии, такие как конвейерные системы или портативные системы транспортирования (например, те- лежки с чистым воздухом, портативные установки с однонаправленным потоком воздуха). Альтернативным решением для защиты частично укупоренных флаконов (например, находящихся в соответствующим образом закрытых боксах) является использование под- донов, закрытых в зоне А и не открываемых повторно в зоне В. Это решение должно быть подтверждено аттестацией,
- Устройства для транспортирования не должны оказывать влияния на потоки воз- духа и вентиляцию зоны загрузки.
- Обращение с негерметизированными контейнерами (такими как частично укупо- ренные флаконы) должно выполняться в зоне А и должно быть, как правило, отделено от операторов физическим барьером или другими достаточными мерами.
- Если укупоривание флаконов пробками не завершается до открывания камеры лиофилизатора, то продукт, удаляемый из лиофилизатора, должен оставаться в зоне А в течение последующего обращения с ним.
- Принадлежности, используемые при загрузке и выгрузке из лиофилизатора (например, поддоны, пакеты, устройства для размещения, пинцеты) должны быть сте- рильными.
Закрытые системы
8.127.Использование закрытых систем позволяет снизить риск загрязнения микроор- ганизмами, частицами и химическими веществами от окружающей среды. Конструкция закрытых систем всегда должна снижать необходимость в ручных манипуляциях и свя- занные с ними риски.8.128.Обеспечение стерильности всех поверхностей, вступающих в контакт с про- дуктом, играет критическую роль для асептического производства. Конструкция и выбор любой закрытой системы для асептического процесса должны обеспечивать сохранение стерильности. Конструкция подсоединения стерильного оборудования (например, трубок и трубопроводов) к тракту движения стерильного продукта после последнего стерилизу- ющего фильтра должна предусматривать асептическое соединение (например, за счет встроенных стерильных соединительных устройств).
8.129.Должны быть приняты необходимые меры для обеспечения целостности ком- понентов, используемых в асептических соединениях. Средства достижения этой цели должны быть определены и указаны в СКЗ. При наличии риска для стерильности продук- та требуется предусмотреть контроль целостности системы. Оценка поставщика должна включать сопоставление данных, относящихся к возможным нарушениям, которые могут привести к потере стерильности системы. 8.130. Окружающая среда, в которой находится закрытая система, должна соответ- ствовать проекту и выполняемому процессу. Для асептического процесса и в случаях лю- бых рисков для нарушения целостности системы, ее следует располагать в зоне А. Если может быть показано сохранение целостности системы при каждом использовании (например, проверкой давления и/или его текущим контролем), то могут использоваться зоны низшего класса. Любые передачи между классифицированными зонами следует тщательно оценивать (п. 4.10). Закрытую систему допускается открывать (например, для технического обслуживания линии производства балк-продукта) в классифицированной зоне, соответствующей материалам (например, зона С для процессов с финишной стери- лизацией или зона А для асептических процессов) или нужно проводить последующую очистку и дезинфекцию (и стерилизацию в случае асептических процессов).Одноразовые системы
8.131.Одноразовые системы в производстве стерильной продукции используются в качестве альтернативы оборудованию многоразового применения. Одноразовые системы могут быть отдельными компонентами или сборками из многих компонентов, таких как контейнеры, фильтры, трубки, соединители, клапаны, бутылки для хранения и сенсоры. Конструкция одноразовых систем должна предусматривать снижение необходимости в манипуляциях и сложности ручных вмешательств. 8.132.Существует ряд факторов риска, специфических для одноразовых систем, ко- торые следует учесть в СКЗ. К этим факторам риска относятся (перечень неисчерпываю- щий):- Взаимодействие между продуктом и поверхностями, вступающими в контакт с продуктом (например, адсорбция, выщелачивание веществ и экстракция).
- Хрупкость системы по сравнению с многоразовыми системами.
- Увеличение числа и сложности ручных операций (включая контроль и обращение с системой) и выполнение соединений.
- Сложностьсборки.
- Проведение контроля целостности до и после использования стерилизующих фильтров (п. 8.87).
- Риск образования проколов и утечек.
- Опасность повреждения системы при открывании внешней упаковки.
- Риск загрязнения частицами.
8.139.Следует обеспечить контроль критических ручных операций с одноразовыми системами, таких как сборка и установка соединений, и проверить их при имитации асеп- тического процесса.
9.Текущий контроль окружающей среды и процессов
Общие положения
9.1.Программа контроля окружающей среды и процесса на производстве является частью общей стратегии контроля загрязнений с целью сведения к минимуму риска за- грязнения частицами и микроорганизмами. Следует учесть, что надежность каждого эле- мента системы текущего контроля (контроль частиц, жизнеспособных организмов и ими- тация асептического процесса) при рассмотрении в отдельности ограничена и их не сле- дует учитывать индивидуально при оценке условий асептики. Их совместное рассмотре- ние позволяет подтвердить надежность проектных решений, аттестации и эксплуатации системы, которые они контролируют. 9.2.Этапрограмма,какправиловключаетвсебяследующие элементы:- Контроль окружающей среды и персонала – общий счет частиц;
- Контроль окружающей среды – жизнеспособные частицы;
- Температура, относительная влажность и другие специальные параметры;
- Имитация асептического процесса (только для продукции, производимой в асеп- тических условиях).
Текущий контроль окружающей среды и процесса
9.4.Следует разработать и документально оформить программу контроля окружаю- щей среды. Целями программы контроля окружающей среды являются:- Обеспечение того, что чистые помещения и устройства очистки воздуха продол- жают поддерживать требуемую чистоту воздуха согласно проекту и нормативным требованиям.
- Эффективное обнаружение отклонений от установленных пределов для окружа- ющей среды, выполняя анализ и оценку влияния на качество продукции.
Следует выполнить оценку рисков для разработки эффективной программы кон- троля окружающей среды, например, определение точек отбора проб, периодичности и методов контроля и условий инкубации (например, времени, температуры, обнаружения аэробных или анаэробных микроорганизмов и др.).
Эта оценка рисков должна быть основана на подробных знаниях о влиянии процесса и готовой продукции, помещениях, оборудовании, критичности конкретных процессов и стадий, выполняемых операциях, данных текущего контроля, полученных прии аттеста- ции данных и данных о типичной микрофлоре, обнаруженной в окружающей среде.
Оценка рисков должна включать определение критических точек контроля, в кото- рых наличие микроорганизмов при выполнении процесса может оказать влияние на каче- ство продукции (например, зоны А, зоны асептического производства и зоны В, которые непосредственно сообщаются с зонами А). Следует учесть и другие данные, такие как ви- зуализация потоков воздуха. Следует регулярно рассматривать оценку риска для под- тверждения эффективности программы текущего контроля окружающей среды на произ- водстве. Программу текущего контроля следует рассматривать в общем контексте анализа тенденций и стратегии контроля загрязнений (СКЗ) на производстве.
9.5.Текущий контроль чистых помещений, устройств очистки воздуха и персонала следует выполнять в эксплуатируемом состоянии на всех критических стадиях производ- ства, включая наладку оборудования. 9.6.Следует контролировать нахождение в заданных пределах других параметров, таких как температура и относительная влажность, согласно требованиям к продукции, процессу и персоналу для обеспечения соответствия стандартам чистоты (например, в зо- нах А или В). 9.7.Контроль зон А должен демонстрировать поддержание асептических условий при выполнении критических операций. Этот контроль следует выполнять в точках с наибольшим риском загрязнения стерильных поверхностей оборудования, первичной упаковки, укупорочных материалов и продукции. Выбор точек контроля, ориентация и расположение пробоотборников должны быть обоснованы, чтобы получить надежные данные для критических зон. 9.8.Методы отбора проб не должны привносить риск загрязнения в процесс произ- водства. 9.9.Следует установить уровни предупреждения и действия для результатов кон- троля частиц и микроорганизмов. Максимальные уровни действия для общего числа ча- стиц приведены в таблице 5, а для жизнеспособных частиц – в таблице 6. Более жесткие пределы для уровней действия могут быть установлены на основе анализа тенденций, особенностей процесса или согласно СКЗ. Уровни предупреждения для общего числа ча- стиц и числа жизнеспособных микроорганизмов следует установить на основе аттестации чистых помещений и периодически рассматриваться с учетом анализа тенденций. 9.10.Следует устанавливать такие уровни предупреждения для зон А (только общее число частиц), зон В, С и D, чтобы обнаруживать и отрицательные тенденции (например, число событий и единичные события, которые указывают на ухудшение параметров окружающей среды. 9.11.Методы текущего контроля должны обнаруживать тенденции изменения пара- метров. К тенденциям относится следующее (перечень неисчерпывающий):- Увеличениечислаотклоненияотуровнейдействияили предупреждения;
- Последующие отклонения от уровней предупреждения;
- Систематические, но единичные отклонения от уровней действия, которые могут иметь общую причину;
- Изменения в видах и количестве микрофлоры и доминирование специфических организмов. Следует обратить особое внимание на повторно появляющиеся организмы, что может указывать на потерю контроля, ухудшение чистоты или трудно контролируе- мые организмы, такие как спорообразующие микроорганизмы и плесени.
Контроль окружающей среды - контроль общего числа частиц
9.14.Следует разработать программу текущего контроля общего числа частиц для получения данных о потенциальных рисках загрязнения и поддержания среды для выпол- нения стерильных операций в аттестованном состоянии. 9.15.В таблице 5 приведены рекомендуемые пределы концентрации аэрозольных ча- стиц при текущем контроле для каждого типа зон.Таблица 5. Максимально допустимые концентрации частиц при текущем кон- троле
|
Зона |
Пределы концентраций частиц с размерами ≥ 0,5 мкм/м3 |
Пределы концентраций частиц с размерами ≥ 5,0 мкм/м3 |
||
|
Оснащенное состояние |
Эксплуатируемое состояние |
Оснащенное состояние |
Эксплуатируемое состояние |
|
|
А |
3 520 |
3 520 |
29 |
29 |
|
В |
3 520 |
352 000 |
29 |
2 930 |
|
С |
352 000 |
3 520 000 |
2 930 |
29 300 |
|
D |
3 520 000 |
Не заданоа) |
29 300 |
Не заданоа) |
а) предел для зон D в эксплуатации не установлен. Производителю следует устано- вить пределы в эксплуатации на основе анализа рисков и данных текущего контроля, если это требуется.
Примечание 1: Приведенные в таблице пределы для концентрации частиц в осна- щенном состоянии должны выполняться после короткого периода «очистки» (рекоменду- емое значение менее 20 мин.), который определяется при аттестации при отсутствии пер- сонала после завершения операций (п. 4.29).
Примечание 2: Случайный счет макрочастиц, особенно с размерами ≥ 5,0 мкм, в зонах А может быть результатом ложных показаний из-за электронных шумов, рассеяния света, ошибки совпадения и др. Однако повторяющийся или систематический счет может указы- вать на возможные загрязнения и требует расследования. Такие события могут служить ранним признаком неисправностей в системах подачи фильтрованного воздуха, отказов оборудования или указывать на небрежность при установке оборудования и эксплуатации.
9.16.Контроль частиц в зонах А следует выполнять в течение всего критического процесса, включая сборку оборудования. 9.17.Контроль зон А следует выполнять для частиц ≥ 0,5 мкм и ≥ 5,0 мкм непрерыв- но с достаточной скоростью отбора проб не менее 28 л/мин (1 фут3/мин) так, чтобы могли быть обнаружены все вмешательства, кратковременные события и любые нарушения в работе системы. Система должна предусматривать частое сравнение результатов отдель- ных проб с уровнями предупреждения и тревоги с периодичностью, при которой могут быть обнаружены возможные отклонения и своевременно предприняты необходимые ме- ры. При превышении предела предупреждения должен быть подан сигнал тревоги. В ин- струкциях должны быть указаны действия в случае тревоги, включая проведение допол- нительного микробиологического контроля. 9.18.Рекомендуется предусматривать такую же систему для зон В, но периодичность отбора проб может быть увеличена. Периодичность контроля зон В и объем пробы долж- ны быть такими, чтобы обнаруживать любое повышение уровня загрязнений или неисправность системы. При превышении предела предупреждения должен быть подан сигнал тревоги. 9.19.Структуру системы мониторинга следует определять на основе анализа рисков, исходящих от используемых в процессе материалов (например, включающих живые орга- низмы, порошки или радиофармпрепараты), которые приводят к повышению биологиче- ской, химической или радиационной опасности. 9.20.Если загрязнения от процесса могут повредить счетчик частиц или представлять опасность (например, живые организмы, порошки или радиация), то частота и метод кон- троля должны быть достаточными для установления класса окружающей среды как до воз- действия фактора риска, так и после него. Следует рассмотреть увеличение объема кон- троля жизнеспособных частиц для обеспечения полного контроля процесса. Текущий кон- троль следует выполнять также при имитирующих операциях через определенные интерва- лы времени согласно системе контроля. Порядок работы должен быть указан в СКЗ. 9.21.Объем пробы при использовании автоматических систем зависит обычно от скорости отбора проб в системе. Объем пробы необязательно должен быть таким же, что для классификации (аттестации) чистых помещений или устройств с чистым воздухом. Объем пробы при контроле должен быть обоснован.Контроль окружающей среды и персонала по жизнеспособным частицам
9.22.В асептическом производстве необходимо достаточно часто проводить микро- биологический контроль с использованием комбинации методов седиментации на чашки, отбора проб в объеме воздуха, с перчаток, одежды и с поверхностей (например, смывы и контактные пластины). Метод отбора проб должен быть обоснован в СКЗ и должно быть показано, что он не оказывает отрицательного влияния на поток воздуха в зонах А и В. Контроль чистых помещений и оборудования следует выполнять в конце работы. 9.23.Следует также контролировать жизнеспособные частицы в чистых помещениях, если обычные производственные операции не выполняются (например, после дезинфек- ции, до начала производства, после завершения выпуска серии продукции и после окон- чания нерабочего периода), и связанных с ними помещениях, которые не использовались, чтобы установить возможные случаи загрязнения, способные повлиять на результаты кон- троля чистых помещений. В случае отклонений может быть выполнен отбор проб в до- полнительных точках для проверки эффективности корректирующих действий. 9.24.В зонах А следует проводить непрерывный контроль (например, отбор проб воздуха или использование седиментационных пластин) в течение всего времени выпол- нения критического процесса, включая сборку оборудования (наладку в асептических условиях) и критические операции. Следует применять аналогичный подход в зонах В, исходя из риска влияния на асептический процесс. Текущий контроль должен охватывать все вмешательства, кратковременные события и сбои в работе, но и сама система кон- троля не должна оказывать отрицательного влияния. 9.25.При оценке риска следует учесть место расположения, вид и частоту контроля исходя из выполняемых действий и близости к критической зоне. В течение процесса сле- дует отбирать пробы с персонала с определенной периодичностью. При отборе проб с персонала не должно быть отрицательного влияния на процесс. Особое внимание следует уделить контролю персонала после выполнения критических операций (как минимум, перчаток, но может потребоваться контроль одежды исходя из особенностей процесса) и при каждом выходе из зоны В (перчатки и одежда). Если контроль перчаток осуществля- ется после выполнения критических операций, то наружные перчатки следует заменять перед продолжением работы. Если контроль одежды осуществляется после выполнения критических операций, то одежду следует заменять перед продолжением работы. 9.26.Следует выполнять микробиологический контроль персонала в зонах А и В. При выполнении ручных операций (например, асептическом смешивании или наполне- нии) микробиологический контроль должен быть усилен с отражением в СКЗ. 9.27.Если текущий контроль выполняется производственным персоналом, то он должен быть предметом регулярных проверок со стороны отдела контроля качества (см. также 8.19). 9.28.Для сокращения времени на контроль микробных загрязнений и снижения рис- ка для продукции производитель может применять альтернативные методы контроля, та- кие как быстрый контроль. Эти быстрые и автоматические методы микробиологического контроля могут применяться после того как их эквивалентность существующим методам или большая эффективность будут показаны при их аттестации. 9.29.Персонал должен полностью понимать методы отбора проб и используемое оборудование. Порядок правильного выполнения операций и оценки результатов должны быть определены инструкциями. Следует иметь вспомогательные данные по эффективно- сти выбранных методов отбора проб.Таблица 6 Максимальные пределы действия для загрязнения жизнеспособны- ми частицами
|
Зона |
В воздухе, КОЕ /м3 |
Седиментационная пластина (диаметр 90 мм), КОЕ/4 часа(а) |
Контактная пластина (диаметр 55 мм), КОЕ/пластинаb) |
Отпечаток перчат- ки, включая 5 паль- цев с обеих рук, КОЕ/перчатка |
|
А |
Нет ростаc) |
|||
|
В |
10 |
5 |
5 |
5 |
|
С |
100 |
50 |
25 |
– |
|
D |
200 |
100 |
50 |
– |
(a) - Седиментационные пластины должны экспонироваться в зонах А и В на протя- жении всего времени выполнения критических операций в течение не более 4 ч (время экспонирования должно быть определено при аттестации с учетом принципа обнаружения микроорганизмов, и не должно оказывать отрицательного влияния на свойства среды).
-
-
- Для зон С и D время экспонирования (максимальная величина 4 ч) и периодичность должны быть основаны на анализе рисков.
- Отдельные седиментационные пластины могут экспонироваться менее 4 ч.
-
(b) Пределы для контактных пластин установлены для поверхностей оборудования, помещений и одежды в зонах А и В. Как правило, не требуется текущий контроль одежды в зонах С и D в зависимости от их работы.
(с) В зонах А ожидаемый результат должен составлять 0 КОЕ. При обнаружении лю- бого роста нужно провести расследование.
Примечание 1: Приведенные в таблице методы контроля являются примерами. Мо- гут использоваться другие методы при условии, что они обеспечивают получение доста- точной информации для всего критического процесса, где продукт может быть загрязнен (например, наладка асептической линии, асептический процесс, наполнение и загрузка в лиофилизатор).
Примечание 2: Во всем документе указаны пределы для КОЕ. При использовании других методов, отличающихся от получения КОЕ, производитель должен научно обос- новать применяемые пределы, и, где возможно, показать соответствие с КОЕ.
9.31.Если в зонах А или В обнаружены микроорганизмы, то следует определить их вид и влияние на качество продукции (для каждой серии, где они обнаружены) и оценить общее состояние контроля. Следует также обратить внимание на идентификацию микро- организмов в зонах C и D (например, если превышены уровни предупреждения и дей- ствия) или после выделения микроорганизмов, которые могут указывать на потерю кон- троля, нарушение условий чистоты или может оказаться затруднительным контроль для таких случаев, как спорообразующие микроорганизмы и плесени с удовлетворительной частотой для понимания в текущем масштабе времени типичной флоры в этих зонах.
Имитация асептического процесса (известная также как «наполнение средами)
9.32.Для периодических проверок эффективности контроля асептических процессов следует проводить имитацию процесса с использованием стерильной питательной среды и/или заменителя (суррогата) вместо продукта. Не следует рассматривать имитацию асеп- тического процесса как основное средство аттестации асептического процесса или частей асептического процесса. Эффективность асептического процесса следует обеспечить за счет проекта процесса, соответствия фармацевтической системе качества и контроля про- цесса, обучения и оценки данных текущего контроля. Выбор питательной среды и/или суррогата должен быть основан на свойстве питательной среды и/или суррогата имитиро- вать физические характеристики продукта исходя из риска для стерильности продукта при выполнении асептического процесса. Если стадии процесса могут косвенно влиять на жизнеспособность любых вносимых микробных загрязнений (например, при производстве в асептических условиях полутвердых продуктов, порошков, твердых материалов, микро- сфер, липосом и других продуктов, когда продукт подлежит охлаждению или нагреву или лиофилизации), то следует разработать альтернативные методы, при которых операции выполняются наиболее близко к реальности. При использовании суррогатов, таких как буферы, в некоторых стадиях имитации асептического процесса, материал суррогата не должен подавлять ростовые свойства потенциальной микрофлоры. 9.33.Имитация асептического процесса должна как можно более точно отражать те- кущий процесс асептического производства и включать в себя все его критические стадии, в особенности:- При имитации асептического процесса следует оценить все асептические опера- ции, выполняемые после стадий стерилизации и деконтаминации материалов, используе- мых в процессе, вплоть до герметизации контейнера;
- Для продукции, не подлежащей фильтрации, следует оценить любые дополни- тельные асептические стадии;
- Если асептический процесс выполняется в инертной атмосфере, то при имитации процесса следует заменить инертный газ на воздух, если только не требуется проведение имитации в анаэробных условиях;
- Для процессов, требующих добавления стерильных порошков, следует использо- вать приемлемый суррогатный материал в тех же контейнерах, что применяются в оцени- ваемом процессе
- Не следует проводить отдельные имитации для отдельных операций (например, сушки, смешивания, измельчения и получения стерильных порошков). Любые индивиду- альные имитации должны быть документально обоснованы и гарантировать, что эти ис- пытания в сумме полностью покрывают весь процесс.
- Имитация процесса для лиофилизированных продуктов должна включать всю цепь асептических операций, включая наполнение, транспортирование, загрузку, предста- вительную длительность выдержки в камере, выгрузку и герметизацию при обоснованных и документированных условиях, соответствующих наихудшему случаю для рабочих па- раметров.
- Имитация процесса лиофилизации должна моделировать все аспекты процесса, кроме тех, которые могут оказать влияние на жизнеспособность или восстановление мик- роорганизмов. Например, следует избегать кипячения или замораживания раствора. При разработке процесса имитации средами следует учесть, где это применимо:
-
- Использование воздуха для сброса вакуума вместо азота или другого техно- логического газа;
- Повторение максимального интервала между стерилизацией лиофилизатора и его использованием;
- Повторение максимального интервала времени между фильтрацией и лиофи- лизацией;
- Количественные аспекты ситуаций наихудшего случая, например, загрузки наибольшего количества поддонов, повторение наибольшей длительности за- грузки, при которой камера открыта в окружающую среду.
- Следует выполнять предусмотренные и корректирующие вмешательства, харак- терные для текущего процесса, таким образом и с такой периодичностью, как они прово- дятся в реальном асептическом процессе.
- Включение вмешательств в асептический процесс и их периодичность должны быть основаны на оценке рисков для стерильности продукции.
- Определение условий наихудшего случая с учетом изменений параметров, таких как размеры контейнеров и скорость конвейера, и их влияние на процесс. Результаты оценки должны обосновывать выбранные параметры.
- Определение представительных размеров комбинаций контейнеров и укупороч- ных материалов для использования при аттестации. При аттестации одних и тех же соче- таний контейнеров и укупорочных материалов для разных видов продукции может быть использован матричный метод (вынесение за скобки), если эквивалентность процесса обоснована научно
- Максимально допустимое время нахождения стерильного продукта и оборудова- ния открытыми в окружающую среду в асептическом процессе.
- Объем наполнения контейнера, достаточный для того, чтобы среда вступала в контакт со всеми поверхностями оборудования и компонентов, которые могут загрязнить стерильный продукт. Объем должен быть таким, чтобы поверхность сверху продукта под- держивала потенциальный рост микроорганизмов и обеспечивала, что при контроле мут- ность может быть определена.
- Требование замены любого инертного газа, используемого в обычном асептиче- ского процессе производства, воздухом, если не предусмотрена имитация с использовани- ем анаэробных микроорганизмов. При таких имитациях следует предусмотреть включе- ние случайных анаэробных имитаций как часть общей стратегии аттестации (п. 9.33, iii).
- Выбранная питательная среда должна поддерживать рост заданной группы кон- трольных микроорганизмов, согласно фармакопее и представителей локальных изолятов.
- Метод определения микробных загрязнений должен быть научно обоснован так, чтобы загрязнения обнаруживались надежно.
- Длительность имитации процесса должна быть достаточной для испытаний про- цесса, операторов, которые выполняют вмешательства, чередования смен и способности окружающей процесс среды создать требуемые условия для производства стерильной продукции.
- Если смены в производстве различаются или имеют увеличенную длительность, то при имитации асептического процесса следует учитывать все специфические для смен факторы, которые представляют риск для стерильности продукции, например, максималь- ное время, в течение которого оператор может находиться в чистом помещении.
- Имитацию прерываний обычного асептического производственного процесса, ко- гда процесс идет в холостую (например, чередование смен, перезагрузка раздаточных со- судов, ввод дополнительного оборудования).
- Удостоверение того, что контроль окружающей среды в текущем производстве выполняется должным образом в течение всей длительности имитации процесса.
- В случае производства циклами, например, при использовании барьерных техно- логий или при производстве стерильных активных субстанций, следует обратить внима- ние на разработку и выполнение имитации процесса так, чтобы при имитации были учте- ны риски, связанные с началом и окончанием цикла и показать, что длительность цикла не приводит ни к какому риску.
- Проведение «имитации асептического процесса в конце производства или цик- ла» может использоваться как дополнительное обоснование или для исследовательских целей; однако это должно быть обосновано в СКЗ и не должно заменять обычные имита- ции асептического процесса. При его выполнении следует показать, что никакой остаточ- ный продукт не оказывает отрицательного влияния на восстановление любого потенци- ального микробного загрязнения.
- Выполнить контроль наполненных единиц при имитации асептического процесса персоналом, прошедшим необходимое обучение на предмет обнаружения микробных за- грязнений. Контроль следует производить в условиях, обеспечивающих идентификацию микробных загрязнений.
- .Пробы наполненных единиц подлежат положительному контролю путем иноку- ляции требуемых контрольных организмов и представительными локальными изолятами.
- Расследование по обнаружению наиболее вероятной первопричины (причин);
- Определить и реализовать требуемые корректирующие действия;
- Выполнить достаточное число успешных последовательных повторных имитаций асептического процесса (обычно не менее трех) чтобы показать, что процесс был возвра- щен в контролируемое состояние.
- В оперативном порядке рассмотреть записи, относящиеся к асептическому произ- водству после последней успешной имитации асептического процесса.
- Результаты рассмотрения должны включать данные по анализу рисков возмож- ных нарушений стерильности в сериях, произведенных после последней успешной имитации асептического процесса;
- В расследование следует включить все другие серии, не выпущенные на рынок. Результаты расследования должны быть учтены при принятии решения об их выпуске
- Данный асептический процесс не выполнялся в течение длительного времени.
- Внесены изменения в процесс, оборудование, инструкции или окружающую сре- ду, которые могут оказать влияние на асептический процесс или добавлены новые комби- нации контейнеров для продукта и укупорочных материалов.
10.Контроль качества
10.1.Предприятие должно располагать персоналом, прошедшим необходимое обуче- ние и имеющим опыт в микробиологии, обеспечении стерильности и знания процессов для выполнения производственной деятельности, контроля окружающей среды и выпол- нения любых расследований по оценке влияния событий, связанных с микробиологией, на безопасность стерильного продукта. 10.2.Спецификации на исходные материалы, компоненты и продукты должны вклю- чать требования к пределам загрязнения микроорганизмами, частицами и эндотоксина- ми/пирогенами, если необходимость в этом следует из данных текущего контроля и/или указаны в СКЗ. 10.3.Для каждой серии продукции следует определять бионагрузку, как для асепти- ческого производства, так и для производства продукции, подлежащей финишной стери- лизации, и результаты следует рассматривать при окончательной оценке серии продук- ции. Следует установить предельные значения загрязнения материала (продукта) непо- средственно перед стерилизующим фильтром или процессом финишной стерилизации в соответствии с используемым методом. Отбираемые пробы должны быть представитель- ными для наихудшего случая (например, в конце работы). Если для продуктов с финиш- ной стерилизацией применяется режим стерилизации «с запасом» (overkill), то бионагруз- ку следует проверять через заданные интервалы времени. 10.4.Для выпуска продукции по параметрам следует разработать дополнительную программу контроля бионагрузки наполненных продуктов до стерилизации. Контроль бионагрузки следует выполнять для каждой серии. Выбор точек отбора проб наполненных единиц до стерилизации должен исходить из наихудшего случая и быть представитель- ным для серии. Следует идентифицировать любые организмы, обнаруженные при микро- биологическом контроле и определить их влияние на эффективность процесса стерилиза- ции. При необходимости следует контролировать уровень эндотоксинов/пирогенов. 10.5.Контроль стерильности готовой продукции следует рассматривать лишь как за- вершающий этап в последовательности мер по обеспечению ее стерильности. Он не мо- жет служить для гарантии стерильности продукции, которая не соответствует параметрам, установленным при разработке, инструкциям или аттестации. 10.6.Контроль стерильности должен выполняться в асептических условиях. Образцы продукции, которые были отобраны для контроля стерильности, должны быть представи- тельными (репрезентативными) для всей серии продукции, особенно для тех частей серии, риск загрязнения которых максимален, например:- Для продуктов, наполнение которыми выполнено в асептических условиях, в ото- бранные образцы должны входить первичные упаковки, наполненные в начале и в конце серии. Следует отобрать дополнительные образцы, например, после любого критического вмешательства, представляющего риск;
- Для продуктов, прошедших тепловую стерилизацию в окончательной упаковке, особое внимание следует уделить отбору образцов в наихудших точках (например, из по- тенциально наиболее холодной или наиболее медленно прогреваемой части загрузки).
- Для лиофилизированной продукции пробы следует отбирать в различных загруз- ках для лиофилизации.
Примечание: Если процесс производства включает подсерии (например, для продук- ции, подлежащей финишной стерилизации), то пробы для контроля стерильности следу- ет отбирать из каждой подсерии и выполнять для каждой подсерии тест на стериль- ность. Следует обратить внимание на отдельное проведение других тестов для готовой продукции.
10.7.Для некоторых продуктов проведение контроля стерильности до выпуска в реа- лизацию может оказаться невозможным из-за слишком короткого срока жизни продукта, не позволяющего завершить контроль стерильности. В этих случаях следует определить и документировать дополнительные меры по разработке процесса и текущему контролю и/или альтернативным методам контроля с целью снижения установленных рисков. 10.8.Любой процесс (например, стерилизации парами перекиси водорода или уль- трафиолетовое облучение), используемый для обработки внешних поверхностей образцов для контроля стерильности перед проведением контроля, не должен оказывать отрица- тельного влияния на чувствительность метода контроля или надежность образца. 10.9.Следует проверять ростовые свойства питательных сред, служащих для кон- троля продукции, по соответствующей фармакопее, до их применения. Ростовые свойства питательных сред, служащих для контроля окружающей среды и имитации асептического процесса, должны быть проверены до их применения с использованием научно обосно- ванных и предназначенных для этого групп микроорганизмов и включая представитель- ные локальные изоляты. Контроль качества питательных сред, как правило, должен вы- полнять конечный пользователь. Использование любых данных контроля сторонними ор- ганизациями, работающими по договору, должно быть обосновано, а условия транспорти- рования и отгрузки должны быть тщательно оценены. 10.10.Данные, полученные при контроле окружающей среды и данные о тенденциях в классифицированных зонах, следует рассматривать при выпуске серии продукции. Сле- дует иметь документально оформленную инструкцию, определяющую действия в случае несоответствия данных контроля окружающей среды тенденции или превышении уста- новленных пределов. Для продукции с коротким сроком жизни данные о параметрах окружающей среды в период производства могут отсутствовать. В этих случаях для под- тверждения соответствия следует рассматривать самые последние полученные данные. Производителям такой продукции следует оценить применение быстрых/альтернативных методов.10.11.При применении методов быстрого и автоматизированного микробиологиче- ского контроля для общих целей производства их следует аттестовать для выпускаемой продукции или процессов.
Термины и определения
Воздушный шлюз (Airlock): небольшое пространство с блокировкой дверей, в кото- ром поддерживается перепад давления между соседними помещениями (как правило, с различными классами чистоты воздуха). Воздушный шлюз предназначен для предотвра- щения проникания частиц и микроорганизмов из зон с более низким классом.
Уровень действия (Action Level): установленный уровень (например, загрязнения микроорганизмами или аэрозольными частицами), после превышения которого требуется провести расследование и предпринять корректирующие действия на основе результатов расследования.
Уровень предупреждения (Alert Level): установленный уровень (например, загряз- нения микроорганизмами или аэрозольными частицами), дающий раннее предупреждение о потенциальном отклонении от нормальных условий эксплуатации и аттестованного со- стояния, по достижении которого не обязательно предпринимаются корректирующие дей- ствия, но проводится анализ для обнаружения потенциальной проблемы. Уровни преду- преждения устанавливаются на основе тенденций при текущем контроле и аттестации и должны периодически пересматриваться. Уровень предупреждения может основываться на ряде факторов, включая отрицательные тенденции, отдельные отклонения от установ- ленных пределов и повторяющихся событий.
Асептическое приготовление/производство (Aseptic preparation/processing) – Обращение со стерильным продуктом, контейнерами и/или устройствами в контролируе- мой окружающей среде, в которой регулируются подготовка воздуха, материалов и пер- сонала для предотвращения загрязнения микроорганизмами, эндотоксинами/пирогенами и частицами.
Имитация асептического процесса (Aseptic Process Simulation – APS) – Имитация всего процесса асептического производства с целью проверки способности процесса обеспечивать стерильность продукции. Включает в себя асептические операции, связан- ные с текущим производством, например, сборку оборудования, приготовление, наполне- ние, лиофилизацию и герметизацию, исходя из необходимости.
Асептические условия (Asepsis): состояние контроля, достигнутое за счет исполь- зования асептической рабочей зоны и выполнения операций с предотвращением микро- биологического загрязнения открытой стерильной продукции.
Тест на удержание бактерий (Bacterial retention testing): этот тест выполняется для подтверждения того, что фильтр способен удалять бактерии из газа или жидкости. Тест обычно выполняется с использованием стандартного организма, такого как Brevundimonas diminuta при минимальной концентрации 107 КОЕ/см2.
Барьер (Barrier): Физическое разделение, которое обеспечивает защиту асептиче- ской технологической зоны (обычно зоны А) за счет разделения с окружающей средой.
Такие системы часто используют частично или полностью барьерные технологии, извест- ные как системы RABS или изоляторы.
Бионагрузка (Bioburden): общее число микроорганизмов, связанных с конкретным объектом, таким как персонал, окружающая производственная среда (воздух и поверхно- сти), оборудование, упаковка продукции, исходные материалы (включая воду), материалы для внутрипроизводственного контроля или готовая продукция.
Биодеконтаминация (Bio-decontamination): Процесс снижения числа жизнеспособ- ных организмов за счет применения спороцидных химических средств.
Примечание переводчика: идентично дезинфекции, см. термин «дезинфекция».
Биологический индикатор (Biological Indicator, BI): популяция микроорганизмов, инокулированных на определенную среду (например, раствор, контейнер или укупороч- ный материал) и помещенная в соответствующие точки загрузки стерилизатора или по- мещения для определения эффективности цикла стерилизации или дезинфекции за счет физического или химического процесса. Контрольный микроорганизм выбирается и атте- стуется исходя из его устойчивости к данному процессу. Качество биоиндикаторов опреде- ляется величиной D , числом микроорганизмов и чистотой.
Технология «выдувание – наполнение – герметизация» (Blow – Fill – Seal, BFS): технология, предусматривающая формование контейнеров из термопластичного грануля- та, наполнение продуктом и герметизацию в условиях непрерывной, цельной, автоматиче- ской операции. Наиболее распространенными являются два вида устройств BFS: челноч- ное устройство (с отрезанием заготовок) и роторное устройство (закрытое отрезание).
Производство циклами (Campaign manufacture) – производство последовательных серий, включающих серии (партии) одного и того же продукта в течение заданного пери- ода времени при строгом соблюдении установленных и аттестованных методов контроля.
Классифицированная зона (Classified area): Зона, включающая ряд чистых поме- щений (см. определение чистого помещения).
Очистка (Cleaning): Процесс удаления загрязнений, например, остатков продукта или дезинфицирующего средства.
Чистая зона (Clean area): зона, в которой установлены требования к чистоте по ча- стицам и микроорганизмам и которая обычно включает в себя ряд сообщающихся чистых помещений.
Чистое помещение (Cleanroom): помещение, разработанное, эксплуатируемое и контролируемое с целью предупреждения загрязнения лекарственных средств частицами и микроорганизмами. Такое помещение имеет заданный класс чистоты, который поддер- живается в эксплуатации.
Классификация чистых помещений (Cleanroom classification): метод оценки чи- стоты воздуха на соответствие спецификации на чистое помещение или оборудование для очистки воздуха путем определения общей концентрации частиц.
Аттестация чистых помещений (Cleanroom qualification): Метод оценки соответ- ствия классифицированного чистого помещения или оборудования для очистки воздуха заданным требованиям.
Закрытая система (Closed system): Система, в которой продукт не открыт в окру- жающую среду. Это, например, может достигаться применением упаковок балк-продукта (например, емкостей или пакетов), которые соединены друг с другом трубками или тру- бопроводами, образуя систему, которая используется для стерильных продуктов, причем система в целом стерилизуется после установки соединений. Примерами могут служить большие системы многоразового использования, такие как в производствах активных суб- станций или одноразовые пакеты и системы с разветвителями в производстве биологиче- ской продукции (перечень неисчерпывающий). Закрытые системы не подлежат открыва- нию до завершения операций. Термин «закрытые системы» в данном приложении не от- носится к таким системам ка RABS и изоляторы.
Колониеобразующая единица, КОЕ (Colony Forming Unit, CFU): микробиологиче- ский термин, относящийся к одной обнаруживаемой колонии, которая произошла из од- ного или более микроорганизмов. Колониеобразующие единицы обычно обозначаются как число КОЕ в 1 мл жидких проб, КОЕ в 1 м3 пробы воздуха для проб, отбираемых на твердую среду, такую как седиментационные или контактные пластины.
Загрязнение (Contamination): Нежелательное внесение примесей микробиологиче- ской природы (количество и тип микроорганизмов, пирогены) или посторонних частиц на или в сырье, активную субстанцию или лекарственное средство при производстве, отборе проб, упаковке или переупаковке, хранении или транспортировании, которые могут ока- зать отрицательное влияние на качество продукции.
Стратегия контроля загрязнений - СКЗ (Contamination Control Strategy – CCS): Плановый комплекс видов контроля микроорганизмов, эндотоксинов/пирогенов и частиц, разработанный на основе понимания выпускаемого продукта или процесса, который обес- печивает выполнение процесса и качество продукции. Контроль может распространяться на параметры и свойства активных субстанций, вспомогательных веществ и материалы лекарственных средств и компонентов, помещения и оборудование, условия работы, внутрипроизводственный контроль, спецификации на готовую продукцию и связанные с ними методы и периодичность контроля, в т. ч. текущего контроля.
Корректирующее вмешательство (Corrective intervention): Вмешательство, выпол- няемое с целью корректировки или регулирования асептического процесса в ходе его вы- полнения. Может не выполняться с установленной периодичностью в ходе асептического процесса. К примерам относятся ликвидация заторов из компонентов, регулировка сенсо- ров и замена частей оборудования.
Критические поверхности (Critical surfaces): поверхности, которые могут вступать в контакт или непосредственно влиять на стерильный продукт, его контейнеры или уку- порочные материалы. Критические поверхности должны быть стерильными до начала производственных операций и поддерживаться стерильными в течение всего процесса.
Критическая зона (Critical Area, Critical zone): зона в асептическом производстве, в которой продукт и критические поверхности открыты в окружающую среду.
Критическое вмешательство (Critical intervention): Вмешательство в критическую зону (корректирующее или предусмотренное).
Величина D (D-value): значение параметра стерилизации (продолжительности или поглощенной дозы), требуемое для снижения числа жизнеспособных специфических мик- роорганизмов в 10 раз от первоначального числа.
Тупик (Deadleg): участок трубы, не являющийся частью циркуляционного контура (в котором жидкость может оставаться неподвижной) и длина которого превышает три внутренних диаметра трубы.
Выведенное из эксплуатации (Decommission): Состояние, когда процесс, оборудо- вание или чистое помещение закрыты и снова использоваться не будут.
Деконтаминация (Decontamination): весь процесс по удалению или снижению лю- бых загрязнений (химических, отходов, остатков или микроорганизмов) из зоны, с объек- та или человека. Следует определить и аттестовать метод деконтаминации (например, очистки, дезинфекции, стерилизации) на предмет достижения требуемого уровня чистоты для предназначенного использования подлежащего деконтаминации объекта. См. также «био-деконтаминация».
Депирогенизация (Depyrogenation): процесс удаления или инактивации пирогенов (например, эндотоксинов) до заданного минимального количества.
Дезинфекция (Disinfection): процесс, при котором снижение числа микроорганиз- мов достигается за счет необратимого действия на их структуру или метаболизм до уста- новленного уровня для данной цели.
Эндотоксин (Endotoxin): пирогенный продукт (например, липополисахарид), при- сутствующий в стенке грамотрицательной бактериальной клетки. Эндотоксин может при- водить к реакциям от воспаления до гибели у пациентов, получивших инъекции.
Время установления равновесия (Equilibration time): Время между достижением температуры стерилизации в контрольной точке измерения и достижением температуры стерилизации во всех точках загрузки.
Экстрагируемые вещества (Extractables): химические соединения, которые мигри- руют с поверхности технологического оборудования, контактирующей с характерными растворителями при экстремальных условиях испытаний, на продукт или материал, участ- вующий в процессе.
Воздух, идущий непосредственно от фильтра (First air): Идущий от фильтра воз- дух, который не прерывается до достижения открытого продукта и поверхностей, вступа- ющих в контакт с продуктом, которые могут добавить загрязнения в воздух до достиже- ния им критической зоны.
Контроль целостности фильтра (Filter Integrity test): Контроль, подтверждающий, что фильтр (для продукта, газа или в системе вентиляции и кондиционирования) сохраня- ет свои свойства по удержанию и не поврежден при обращении, монтаже или при выпол- нении процесса.
«Формование–наполнение–герметизация» (Form–Fill–Seal - FFS): автоматизиро- ванный процесс наполнения, используемый обычно для продуктов, подлежащих финиш- ной стерилизации, при котором первичный контейнер формируется из непрерывного плоского рулона упаковочной пленки. При этом одновременно происходит наполнение полученного контейнера продуктом и герметизация наполненного контейнера в непре- рывном процессе. В процессе «формование–наполнение–герметизация» может использо- ваться система c одним полотном (когда контейнер образуется оборачиванием одного ру- лона с плоской пленкой вокруг себя) или система с двумя полотнами (когда два рулона с плоской пленкой соединяются для образования контейнера), при этом часто используются вакуумная сварка или газы под давлением. Полученный контейнер наполняется, гермети- зируется и разрезается на секции. Пленка обычно состоит из полимерного материала, по- крытой пленкой фольги или другого подходящего материала.
Аттестация переодевания (Gowning Qualification): программа, которая устанавли- вает порядок проверки способности индивидуума надевать комплект стерильной одежды; предусматривается первоначальная и периодические проверки.
Воздух, соответствующий зоне А (Grade A air supply): воздух, который прошел че- рез фильтр, аттестованный на способность получения воздуха для зоны А по общему чис- лу частиц, но где нет требований по непрерывному контролю общего числа частиц или соответствия зоне А по пределам для жизнеспособных частиц. Этот термин используется для защиты полностью укупоренных флаконов до закатки колпачков.
НЕРА фильтр (HEPA filter): высокоэффективный фильтр очистки воздуха по части- цам в соответствии принятым международным стандартом.
Предусмотренное вмешательство (Inherent intervention): вмешательство, которое является составной частью асептического процесса и требуется для наладки, текущей ра- боты и/или контроля (например, асептическая сборка, повторное наполнение контейнеров, отбор проб из окружающей среды). Может быть предусмотрено технологией или рабочей инструкцией для выполнения асептического процесса.
Встроенное стерильное соединительное устройство (Intrinsic sterile connection device): устройство, которое уменьшает риск загрязнения в процессе соединения; оно мо- жет быть механическим или паяным.
Изокинетический пробоотборник (Isokinetic sampling head): пробоотборник, кото- рый вносит минимально возможные нарушения в поток воздуха так, что в его отверстие попадают те же частицы, которые прошли бы через площадь отверстия при отсутствии пробоотборника (т. е. условия отбора проб, при которых средняя скорость воздуха, вхо- дящего в пробоотборник примерно равна ± 20% средней скорости воздуха в этой точке).
Изолятор (Isolator): устройство, внутри которого может выполняться деконтамина- ция, с внутренней рабочей зоной, соответствующей требованиям к зоне А, что обеспечи- вает полную и непрерывную изоляцию внутреннего объема от внешней окружающей сре- ды (например, воздуха окружающего чистого помещения и персонала). Существует два основных типа изоляторов:
Закрытые изолирующие системы – исключают внешнее загрязнение внутреннего объема изолятора за счет организации передачи материалов через асептическое соедине- ние вспомогательному оборудованию, а не за счет использования отверстий в окружаю- щую среду. Закрытые системы остаются герметичными в течение всего времени выполне- ния операции.
Открытые изолирующие системы – предусматривают непрерывную или полуне- прерывную подачу и/или выдачу материалов при проведении операций через одно или более отверстий. Отверстия устроены так, чтобы исключить попадание загрязнений из наружной среды в изолятор (например, за счет непрерывного избыточного давления).
Выщелачиваемые вещества (Leachables): химические соединения, которые мигри- руют в лекарственные средства из поверхности технологического оборудования, контак- тирующей с продуктом, или контейнеров при нормальных условиях использования и/или хранения.
Локальные изоляты (Local isolates): Характерные для данного производства пред- ставительные микроорганизмы, которые часто обнаруживаются при контроле окружаю- щей среды в пределах классифицированной зоны (зон), особенно в зонах А и В, контроле персонала или результатах положительного контроля на стерильность.
Лиофилизация (Lyophilization): физико-химический процесс высушивания, в кото- ром происходит удаление растворителей как из водных, так и из неводных систем, пред- назначенный для обеспечения стабильности продукта или материала. Синонимом термина
«лиофилизация» является «сублимационная сушка».
Ручной асептический процесс (Manual aseptic processing): асептический процесс, при котором оператор выполняет вручную приготовление, наполнение, размещает и/или герметизирует открытый контейнер со стерильным продуктом.
Оператор (Operator): физическое лицо, участвующее в выполнении технологических операций, включая наладку, наполнение, техническое обслуживание или другой персонал, связанный с производственной деятельностью.
Стерилизация «с запасом» (Overkill sterilization): процесс, предусматривающий снижение популяции микроорганизмов не менее чем в 1012 раз при минимальной вели- чине D, равное 1 мин.
Заготовка для контейнера (Parison): полимерная «трубка», экструдированная из установки «выдувание-наполнение-герметизация»), из которой формуется контейнер.
Передаточная камера (Pass-through hatch): синоним термину «воздушный шлюз», но, как правило, меньшая по размерам.
Пациент (Patient): Человек или животное, участвующее в клинических испытаниях.
Финишная обработка теплом после асептического процесса (Post-aseptic pro- cessing terminal heat treatment): Финишная обработка влажным теплом, выполняемая после асептического процесса, для которой показана способность обеспечивать гарантирован- ный уровень стерильности ≤ 10-6, но где требования к стерилизации паром (например, ве- личина F0 ≥ 8 мин) не выполнены. Это может способствовать деструкции вирусов, кото- рые могут не удалены путем фильтрации.
Пироген (Pyrogen): вещество, вызывающее фебрильную реакцию (повышение тем- пературы) пациента после инъекции.
Система/порт для быстрой передачи (Rapid Transfer System/Port – RTP): Система, служащая для передачи предметов в устройство RABS или изоляторы, которая сводит к минимуму риск для критической зоны. Примером может служить альфа/бета порт.
Исходный материал (Raw material): Любой ингредиент, предназначенный для ис- пользования в производстве стерильной продукции, включая те, которые отсутствуют в готовом лекарственном средстве.
Барьерная система с ограниченным доступом (Restricted Access Barrier System, RABS): устройство, отделенное, но не полностью герметизированное, внутренний объем которого соответствует заданным требованиям к чистоте воздуха (зоне А для асептиче- ских процессов), и имеющее оболочку из твердых стенок и встроенные перчатки для от- деления внутренней среды от окружающего чистого помещения. Внутренние поверхности RABS подлежат дезинфекции или деконтаминации спороцидным средством. Операторы используют перчатки, полукостюмы, RTP или другие встроенные передаточные порты для выполнения манипуляций или передачи материалов внутрь RABS. В зависимости от конструкции, двери открываются редко и только при строго определенных условиях.
Одноразовые системы (Single Use Systems, SUS): системы, в которых контактиру- ющие с продуктом элементы используются только один раз, заменяющие оборудование повторного использования, такое как передающие линии из нержавеющей стали или балк- контейнеры. Для целей данного руководства, к одноразовым системам относятся системы, используемые в процессах производства стерильной продукции и обычно изготовленные из одноразовых компонентов, таких как мешки, фильтры, трубки, соединения, емкости для хранения и сенсоры.
Спороцидное средств (Sporicidal agent): Средство, которое разрушает споры бакте- рий и грибов при использовании в достаточных концентрациях в течение заданного вре- мени контакта. Предполагается, что оно убивает все вегетативные микрооргнизмы.
Стерильная продукция (Sterile Product): для целей данного руководства под сте- рильной продукцией понимается один или несколько стерильных элементов, открытых в асептическую среду и в итоге образующих стерильную активную субстанцию или сте- рильный готовый продукт. К этим элементам относятся контейнеры, укупорочные мате- риалы и компоненты готового лекарственного средства. К ней относится также продукт, который считается стерильным после финишной стерилизации.
Стерилизующий фильтр (Sterilizing grade filter): фильтр, который после проведения аттестации удаляет определенное количество микроорганизмов из потока жидкости или газа, образуя стерильный поток. Обычно такие фильтры имеют размер пор, равный или меньший 0,22 мкм.
Финишная стерилизация (Terminal sterilization): процесс, при котором воздействие стерилизующего агента или условий в летальной дозе на продукт в окончательном кон- тейнере обеспечивает заранее определенный гарантированный уровень стерильности 10-6 или лучше (т. е. теоретическая вероятность выживания микроорганизма в стерилизован- ной единице продукции равна или меньше 10-6, или 1 единица из миллиона).
Турбулентный поток воздуха (Turbulent airflow): Поток воздуха, не являющийся однонаправленным. В чистом помещении турбулентный поток воздуха должен обтекать помещения с разбавлением смешанного потока и поддержания приемлемого качества воз- духа.
Однонаправленный поток воздуха (Unidirectional airflow): поток воздуха, движу- щийся в одном направлении, отвечающий условиям устойчивости и однонаправленности и имеющий достаточную скорость для непрерывного удаления частиц из критической технологической или контрольной зоны.
Устройство с однонаправленным потоком воздуха (Unidirectional Airflow (UDAF) unit): Устройство, в которое подается прошедший фильтр однонаправленный поток возду- ха (ранее называемое устройством с ламинарным потоком).
Наихудший случай (Worst case): комплекс условий, включающий пределы для про- цесса и обстоятельства, в том числе содержащиеся в инструкциях, при которых вероят- ность нарушения процесса или продукта (в сравнении с идеальными условиями) будет наибольшей. Такие условия представляют риск, но не обязательно приводят к нарушени- ям процесса или продукта.
Система подготовки воды (Water system): Система для приготовления, хранения и распределения воды, обычно в соответствии требованиями фармакопеи (например, вода очищенная и вода для инъекций).
Величина z (Z-value): Разность температур, при которой величина D изменяется в 10 раз для биологического индикатора.
Область применения
- Настоящее приложение устанавливает требования к обращению с контрольными образцами исходных, упаковочных материалов, готовой продукции и с архивными образцами готовой продукции.
- Специальные требования к лекарственным средствам для исследований, приведены в приложении 13 к настоящему стандарту.
- Настоящее приложение также распространяется на работу с архивными образцами лекарственных средств, реализуемых (импортируемых) несколькими дистрибьюторами.
2 Общие положения
- Образцы следует хранить для проведения:
- аналитических исследований;
- анализа готовой продукции в случае необходимости.
- Производитель, импортер и/или субъект, выпускающий серию продукции (см. пункты 7 и 8 данного приложения), должны хранить контрольные и/или архивные образцы от каждой серии готовой продукции. Производитель также должен хранить контрольные образцы от каждой серии исходных материалов (кроме исключений, приведенных в пункте 3.2 данного приложения) и/или промежуточной продукции. На каждом участке по упаковке следует хранить контрольные образцы от каждой серии первичных упаковочных материалов и печатных материалов. Допускается включать печатные материалы в состав контрольных и/или архивных образцов готовой продукции.
- Контрольные и/или архивные образцы характеризуют серию готовой продукции или исходных материалов, являются приложением к протоколу серии и могут быть оценены в случае, например, рекламаций на качество лекарственного средства, проверке соответствия требованиям, установленным при государственной регистрации, проверке маркировки и упаковки или при проверке надзорными органами (инспекцией).
- Следует вести документацию, позволяющую проследить происхождение образцов, и представлять ее в надзорные органы.
3 Срок хранения
- Контрольные и архивные образцы от каждой серии готовой продукции следует хранить, как минимум, в течение срока годности серии и одного года после истечения срока годности. Контрольный образец должен быть упакован в его первичную упаковку или в упаковку, состоящую из того же материала, что и первичная упаковка, в которой препарат реализуется на рынке (указания в отношении импортируемых лекарственных средств для животных, кроме иммунных препаратов, даны в пунктах 8 и 9 приложения 4 к настоящему стандарту).
- Образцы исходных материалов (кроме растворителей, газов или воды, предназначенных для технологических целей) должны храниться в течение не менее двух лет после выпуска продукции, если более длительный период не предусмотрен нормативными документами. Это время может быть сокращено, если в спецификации указан более короткий период стабильности материала. Упаковочные материалы должны храниться в течение срока хранения соответствующего готового продукта.
Правила GMP EC 2019
4 Количество контрольных и архивных образцов
- Количество контрольных и архивных образцов должно быть достаточным для проведения не менее двукратного аналитического контроля серии продукции в соответствии с требованиями, установленными при государственной регистрации. В случае необходимости следует для каждого вида аналитического контроля использовать невскрытые упаковки. Любые исключения должны быть обоснованы и согласованы с соответствующим органом.
- Необходимо соблюдать требования в отношении количества контрольных образцов и, при необходимости, архивных образцов.
- Контрольные образцы должны быть представительными для серии исходных материалов, промежуточной или готовой продукции, из которой они отобраны. Для контроля наиболее критических этапов процесса (например, начала или конца процесса) могут отбираться дополнительные образцы. Если процесс упаковки серии ведется в ходе двух и более операций по упаковке, то после каждой из этих операций следует отбирать не менее одного архивного образца. Любые исключения должны быть обоснованы и согласованы с соответствующим органом.
- Все необходимые материалы и оборудование для проведения контроля в соответствии со спецификацией должны быть в наличии (или быть легко доступными) до истечения срока годности последней выпускаемой серии и одного года после истечения срока годности.
5 Условия хранения
- Хранение контрольных образцов готовой продукции и активных субстанций должно быть организовано в соответствии с требованиями нормативных документов на лекарственные средства и активные субстанции.
- Условия хранения должны соответствовать требованиям, установленным при регистрации лекарственного средства (например, требований к пониженной температуре, если требуется).
6 Письменные соглашения
- Если держатель лицензии на производство не является одновременно юридическим лицом, ответственным за выпуск серии продукции в Российской Федерации*, обязанность по отбору и хранению контрольных/архивных образцов должна быть определена в письменном соглашении между двумя сторонами в соответствии с разделом 7 части I данного стандарта. Это также касается случаев, когда какая-либо деятельность по производству или выпуску серии продукции проводится не на том предприятии (производстве), которое несет общую ответственность за серию продукцию на рынке Российской Федерации*. Порядок отбора и хранения контрольных и архивных образцов для каждого предприятия (производства) должен быть определен в письменном соглашении.
- Уполномоченное лицо, которое выдает разрешение на выпуск, должно убедиться в том, что все контрольные и архивные образцы будут в наличии в течение установленного периода. При необходимости порядок получения образцов должен быть определен в письменной форме.
- Если в производстве готовой продукции принимает участие более одного предприятия (производства), то порядок отбора и места хранения контрольных и архивных образцов должны быть определены в письменной форме.
7 Контрольные образцы. Общие положения
- Контрольные образцы предназначены для проведения анализа и должны быть доступны для лаборатории, имеющей аттестованные методики его проведения. Образцы исходных материалов, используемых в производстве лекарственных средств в Российской Федерации*, и образцы готовой продукции должны храниться на предприятии-производителе готовых лекарственных средств.
- Порядок обращения с контрольными образцами готовых лекарственных средств, производимых в других странах:
- Если страна имеет соглашение о взаимном признании с Российской Федерацией*, то контрольные образцы могут отбираться и храниться на предприятии-производителе. Это должно быть оформлено письменным соглашением между импортером внутри Российской Федерации и производителем, находящимся за ее пределами.
- Если страна не имеет соглашения о взаимном признании с Российской Федерацией, то контрольные образцы готовой продукции следует отбирать и хранить на уполномоченном предприятии, расположенном в Российской Федерации. Отбор образцов должен выполняться в соответствии с письменным соглашением между всеми сторонами. Рекомендуется хранить образцы там, где проводился контроль продукции при ее ввозе.
- В оригинале правил GMP ЕС указано ЕС (прим. разработчика стандарта).
Правила GMP EC 2019
- Контрольные образцы исходных и упаковочных материалов следует хранить там, где они использовались для производства лекарственных средств.
8 Архивные образцы
Общие положения
- Архивные образцы должны быть представительными для серии готовой продукции, реализуемой в Российской Федерации, и использоваться для контроля с целью подтверждения соответствия требованиям, установленным при государственной регистрации, или требованиям законодательства Российской Федерации (исключая технические параметры). В связи с этим архивные образцы должны храниться в пределах Российской Федерации. Рекомендуется хранить их в месте нахождения уполномоченного лица, выдавшего разрешение на выпуск продукции.
- Если действует соглашение о взаимном признании, и контрольные образцы хранятся у производителя, находящегося в стране за пределами Российской Федерации (см. пункт 7.2.2 данного приложения), отдельные архивные образцы должны храниться в Российской Федерации (см. пункт 8.1 данного приложения).
- Архивные образцы должны находиться у производителя, имеющего лицензию, и быть доступными для представителей надзорных органов.
- Если в последовательности «ввоз – процесс упаковки – контроль – выпуск серии» участвует более одного производителя в пределах Российской Федерации, то ответственность за отбор и хранение архивных образцов должна быть определена письменным соглашением между участвующими сторонами.
9 Контрольные и архивные образцы продукции, импортируемой (реализуемой) несколькими дистрибьюторами
- Если вторичную упаковку лекарственного средства не вскрывают, то следует хранить только используемый упаковочный материал, поскольку риск перепутывания продукции низок или отсутствует.
- Если вторичную упаковку вскрывают, например, для замены картонной коробки или листкавкладыша, то следует отбирать один архивный образец для каждой операции процесса упаковки, т. к. существует риск перепутывания продукции в процессе упаковки. Следует предусмотреть порядок, позволяющий быстро определять виновного в перепутывании (производитель или дистрибьютор), т. к. от этого зависит объем отзываемой продукции.
10 Контрольные и архивные образцы в случае ликвидации предприятияпроизводителя
- В случае ликвидации предприятия-производителя и отзыва (отмены, истечения срока действия) лицензии на производство на рынке может остаться большое количество серий продукции с не истекшим сроком годности. В этом случае производитель обязан передать контрольные и архивные образцы (и соответствующую документацию) на хранение в предназначенное для этого место. Производитель должен обосновать перед надзорным органом достаточность принятых мер по хранению и возможность передачи образцов для проведения оценки и анализа, при необходимости.
- Если производитель не может сделать этого, то выполнение необходимых действий может быть передано другому производителю. Держатель лицензии на производство несет ответственность за такую передачу функций и за представление необходимой информации надзорному органу. Кроме того, он должен согласовать с надзорным органом достаточность мер по хранению контрольных и архивных образцов.
- Эти требования распространяются также на случай ликвидации производства, находящегося за пределами Российской Федерации. В этом случае импортер несет ответственность за принятие необходимых мер и согласование с соответствующими органами.
Область применения
Настоящее приложение устанавливает порядок подтверждения соответствия, выполняемого уполномоченным лицом, и требования к выпуску серий лекарственных средств для человека и животных в реализацию согласно действующему порядку. Принципы данного приложения применяются также к лекарственным средствам для исследований, предназначенных для человека. Настоящее приложение не охватывает все возможные случаи подтверждения соответствия и не относится к продукции, выпуск которой регламентируется специальными требованиями (такими, как препараты крови или иммунные препараты). Основные требования к выпуску серии продукции приводятся в регистрационном досье и документах, оформленных при государственной регистрации. Ни одно из положений настоящего приложения не может использоваться, если оно выходит за рамки установленных требований.Основные положения
Держатель регистрационного досье несет полную ответственность за характеристики лекарственного средства в течение его срока годности, за его безопасность, качество и эффективность. Уполномоченное лицо несет ответственность за подтверждение того, что каждая отдельная серия продукции произведена и проверена в соответствии действующими требованиями, регистрационным досье и настоящим стандартом (GMP). Процесс выпуска серии включает в себя следующее:- Проверку производства и контроля серии в соответствии с инструкцией по выпуску продукции.
- Сертификацию серии готовой продукции уполномоченным лицом, удостоверяющим, что серия соответствует требованиям настоящего стандарта (GMP) и регистрационному досье. Это является подтверждением качества выпускаемой серии.
- Передачу ан склад готовой продукции (торговый склад) и/или отправку на экспорт серии готовой продукции, которые следует выполнять с учетом сертификации уполномоченным лицом. Если эта передача осуществляется не на площадке, на которой выполнена сертификация, то ее следует проводить в соответствии с письменным соглашением между сторонами.
- Серия произведена и проверена в соответствии с требованиями регистрационного досье.
- Серия произведена и проверена в соответствии с требованиями настоящего стандарта (GMP).
- Учтены все другие относящиеся к предмету требования.
- В случае, если требуется расследование или отзыв согласно главе 8 части I настоящего стандарта, должны быть доступны уполномоченное лицо, выполнившее сертификацию или подтверждение1, и вся необходимая документация.
1 Процесс сертификации
- Каждая серия готовой продукции должна быть сертифицирована2 уполномоченным лицом до ее выпуска в реализацию или отправки на экспорт. Сертификация может быть выполнена только уполномоченным лицом производителя и/или импортера, указанным в регистрационном досье.
- Уполномоченное лицо, выполняющее сертификацию или подтверждение серии, должно четко знать область своей ответственности. Уполномоченное лицо должно периодически проходить обучение в отношении вида продукции, процесса производства, технического развития и изменений в настоящем стандарте (GMP).
- Различные стадии процесса производства серии до сертификации, импорта, испытаний и хранения могут выполняться на разных площадках. Независимо от числа площадок уполномоченное лицо, выполняющее сертификацию готовой продукции, должно удостовериться в том, что все необходимые стадии выполнены в соответствии с фармацевтической системой качества согласно настоящему стандарту (GMP), регистрационному досье и другим требованиям.
- Каждая производственная площадка должна располагать, как минимум, одним уполномоченным лицом.
- Если на площадке выполняется лишь часть производственных операций с серией, то уполномоченное лицо на этой площадке должно, кк минимум, подтвердить, что операции на площадке выполнены в соответствии с настоящим стандартом (GMP) и письменным соглашением, в котором подробно указаны операции, за которые отвечает данная площадка. Если уполномоченное лицо отвечает за подтверждение соответствия этих операций регистрационному досье, то оно должно иметь доступ ко всем материалам досье.
- Уполномоченное лицо, которое сертифицирует серию готовой продукции, может нести полную ответственность за все стадии производства серии или эта ответственность может быть разделена с другими уполномоченными лицами, подтверждающими отдельные стадии производства и контроля серии. Это могут быть другие уполномоченные лица, которые работают у одного и того же держателя регистрационного досье, либо у разных держателе регистрационного досье.
- Любое разделение ответственности между уполномоченными лицами в отношении подтверждения серии определено в документе, согласованном всеми сторонами. Этот документ должен детально определять ответственность за оценку влияния любых отклонений серии от требований настоящего стандарта (GMP) или регистрационного досье.
- Для лекарственных средств, произведенных за пределами Российской Федерации, ввоз продукции и сертификация являются завершающими стадиями производства, после которых серия передается на торговый склад.
- Порядок сертификации приведен в разделе 1 данного приложения и распространяется на все лекарственные средства, предназначенные для реализации в Российской Федерации или для экспорта, независимо от сложности цепи снабжения и расположения производств.
- В соответствии с п. 1.4. данного приложения уполномоченное лицо, выполняющее сертификацию серии готового лекарственного средства, может учесть подтверждение от других уполномоченных лиц, разделяя определенную ответственность в отношении производства или импорта, осуществляемого на других площадках в Российской Федерации или другими держателями регистрационного досье.
- Уполномоченное лицо должно учитывать условия хранения и транспортирования серии и образцов, если они отправляются раздельно, до выдачи сертификата на серию.
- Уполномоченное лицо, сертифицирующее готовую продукцию, несет ответственность за то, что каждая серия готового лекарственного средства произведена в соответствии с требованиями настоящего стандарта (GMP) и регистрационного досье. За исключением случаев, когда между Российской Федерацией и экспортирующей страной заключено соглашение о взаимном признании инспекций или аналогичное соглашение, уполномоченное лицо отвечает за проведение в полном объеме количественного и качественного анализа, как минимум, для всех активных субстанций и всех других испытаний или проверок, необходимых для подтверждения качества лекарственных средств в соответствии с регистрационным досье.
- Отбор образцов импортируемой продукции должен быть полностью представительным для серии. Образцы могут отобраны при ввозе продукции в Российскую Федерацию или отобраны на производственной площадке в другой стране в соответствии техническим обоснованием, которое входит в систему качества компании. Ответственность за отбор образцов должна быть определена в письменном соглашении между сторонами. Порядок транспортирования образцов, отобранных за пределами Российской Федерации, должен соответствовать требования к транспортированию серии, которую они представляют.
- Если отбор образцов выполняется на производственной площадке в другой стране, то техническое обоснование должно включать в себя формальную оценку риска для установления и предотвращения рисков. Эта оценка должна быт оформлена документальна и включать, как минимум, следующее:
- Аудит производственной деятельности, включая отбор образцов в другой стране, и оценка последующих этапов транспортирования серии и образцов для подтверждения того, что образцы являются представительными для ввозимой серии.
- Детальный научный анализ, включая анализ вспомогательных данных для подготовки заключения о том, что образцы отобранные в другой стране, являются представительными для ввозимой серии. Этот анализ должен включать, как минимум:
- Описание процесса отбора образцов в другой стране;
- Описание условий транспортирования образцов и ввезенной серии с обоснованием любых отклонений;
- Сравнительный анализ образцов, отобранных в другой стране и образцов, отобранных после ввоза;
- Длительность интервала времени между отбором образцов и ввозом серии и подготовка данных для обоснования пределов.
- Проведение рандомизированного периодического анализа образцов, отобранных после ввоза, для обоснования доверия к образцам, отобранным в другой стране.
- Рассмотрение любых необычных результатов или обнаруженных отклонений от спецификации. Это следует учесть при оценке доверия к образцам, отобранным на производственной площадке в другой стране и о результатах следует уведомлять надзорный орган в введении которого находится площадка, где проводится сертификация. Такое событие следует рассматривать как потенциальный дефект для качества и расследовать согласно главе 8 части I настоящего стандарта.
- Различные серии ввозимой готовой продукции могут быть произведены из одной серии нерасфасованной продукции (балк-продукции). Уполномоченные лица, выполняющие сертификацию разных серий готовой продукции могут основывать свои решения на контролю качества первой ввезенной серии готовой продукции при условии документального обоснования использованием анализа рисков. При этом следует учесть требования п. 1.5.6 в отношении доверия к образцам, отобранным в других странах. Обоснование подтверждения целостности и идентичности серии ввезенной готовой продукции должно быть оформлено документально и включать как минимум, следующее:
- Требования к хранению нерасфасованной продукции (балк-продукции) до расфасовывания;
- Условия хранения и транспортирования серии готовой продукции соответствуют установленным сериям;
- Груз находится с сохранном состоянии и отсутствуют признаки его повреждения при хранении или транспортировании;
- Обозначение продукции выполнено правильно;
- Испытанные образцы являются представительными для всех серий готовой продукции, полученной из одной серии нерасфасованной продукции.
- Уполномоченное лицо должно лично удостовериться в выполнении следующих условий до выдачи сертификата на серию для выпуска в реализацию или для экспорта:
- Сертификация выполнена в соответствии с регистрационным досье;
- Выполнены другие требования в соответствии с законодательством;
- Сертификат зарегистрирован в соответствующем регистре или другом эквивалентом документе.
- В дополнение к этому уполномоченное лицо отвечает за выполнение требований п.п. 7.1–
- Вся деятельность по производству и контролю лекарственных средств должна соответствовать настоящему стандарту (GMP).
- Вся цепь поставок активной субстанции и лекарственного средства вплоть до сертификации должна быть оформлена документальная и быть доступна уполномоченному лицу. К этому относятся проа исходных и упаковочных материалов для лекарственного средства и любые другие материалы, отнесенные к критическим по результатам анализа рисков в производственном процессе. Документ предпочтительно оформит в виде схемы, на которой показаны все участвующие стороны, включая субподрядчиков для выполнения критических стадий, например, стерилизации материалов и оборудования для асептического производства.
- Проветение всех аудитов площадок, участвующих в производстве и контроле лекарственных средств и производстве активных субстанций и все отчеты об аудитах доступны уполномоченному лицу, проводящему сертификацию.
- Все площадки по производству, проведению анализов и сертификации соответствуют требованиям регистрационного досье.
- Вся деятельность по производству и контролю соответствует требованиям регистрационного досье.
- Источник и спецификации на исходные и упаковочные материалы, использованные в производстве серии, соответствуют регистрационному досье. Поставщик имеет систему качества, которая предусматривает поставку только материалов требуемого качества.
- Лекарственные средства и активные субстанции для них произведены в соответствии с настоящим стандартом (GMP), и где требуется, их реализация выполняется в соответствии с правилами обращения в системе оптовых поставок (GDP) для активных субстанций.
- Ввоз активных субстанций, используемых для производства лекарственных средств для человека, соответствует действующим требованиям.
- Вспомогательные вещества для лекарственных средств, на которые распространяется настоящий стандарт, также произведены в соответствии с ним.
- Где требуется, статус по трансмиссивной губчатой энцефалопатии всех материалов, используемых в производстве серии соответствует регистрационному досье.
- Вся документация находится в полном составе и утверждена лицами, имеющими на то право. Все виды внутрипроизводственного контроля и проверки выполнены.
- Все процессы производства и контроля остаются в аттестованном состоянии. Персонал прошел обучение и имеет требуемую квалификацию.
- Данные контроля качества готовой продукции соответствуют спецификации на готовую продукцию согласно регистрационному досье или, где это разрешено, программе выпуска в реальном времени.
- Все обязанности перед надзорными органами после реализации, относящиеся к производству или контролю, выполнены. Последующий анализ стабильности подтверждает сертификацию.
- Проведена оценка влияния любых факторов на производство или контроль и выполнены дополнительные проверки и испытания.
- Все исследования, относящиеся к сертифицируемой серии (включая расследования причин выхода за пределы спецификации и необычных тенденций), завершены с результатами, достаточными для сертификации.
- Любые последующие рекламации, расследования или отзывы не влияют на условия сертификации данной серии.
- Существует необходимое техническое соглашение.
- Действует и поддерживается программа самоинспекций.
- Действует порядок сбыта и отгрузки.
- При поставке лекарственных средств для человека на рынок Российской Федерации на упаковках предусмотрены знаки (средства) безопасности в соответствии с требованиями, если это необходимо.
- Для некоторых видов продукции могут применяться специальные руководства, например, приложение 2 к настоящему стандарту «Производство биологически активных субстанций и лекарственных средств для человека» и приложение 3 «Производство радиофармацевтических препаратов».
- В случае параллельного импорта и параллельной поставки любые операции по переупаковке уже выпущенной серии должны быть согласованы с надзорным органом.
- До сертификации переупакованной серии уполномоченное лицо должно подтвердить соответствие действующим требованиям к параллельному импорту и параллельной поставе.
- Уполномоченное лицо держателя регистрационного досье, ответственное за сертификацию серии переупакованной готовой продукции, должно удостоверить, что переупаковка выполнена с требованиями, относящимися к переупакованной продукции и настоящему стандарту (GMP).
- Регистрация сертификации уполномоченным лицом.
- Сертификация лекарственного средства регистрируется уполномоченным лицом в регистре или эквивалентном документе, предназначенном для этой цели. Запись о регистрации должна свидетельствовать, что каждая серия соответствует действующим требованиям. Запись должна быть сделана после выполнения операции и сохраняться в течение установленного периода времени, но не менее пяти лет.
- Контрольный отчет должен быть выполнен в соответствии с действующими требованиями.
2 Использование оценок соответствия требованиям GMP, выполненных третьими сторонами, например, аудитов
В некоторых случаях уполномоченное лицо может доверять правильно функционирующей фармацевтической систем качества производств, выпускающих продукцию, и основываться на аудитах, проведенных третьими сторонами.- Доверие оценкам, выполненными третьими сторонами, должно основываться на главе 7 части I настоящего стандарта (GMP) для того, чтобы определить, согласовать и контролировать работу по контракту.
- Особое внимание следует уделить согласованию отчетов о проведении аудитов:
- Отчет о проведении аудита должен отражать соответствие требованиям настоящего стандарта (GMP), например, в отношении фармацевтической системы качества, всей документации по производству и контролю продукции, например, производству активных фармацевтических субстанций, контролю качества, первичной упаковке и т. д. Все охваченные аудитом стороны работы должны быть четко изложены и оформлены в виде отчета о проведении аудита.
- Следует установить, соответствует ли производство и контроль качества активной субстанции и лекарственного средства требованиям GMP, или, в случае производства в третьей стране, являются ли действующие в ней правила GMP эквивалентными настоящему стандарту.
- В случае работы по контракту следует проверить ее соответствие регистрационному досье.
- Уполномоченное лицо должно убедиться в том, что выполнена оценка и согласование отчета о проведении аудита третьей стороной. Уполномоченное лицо должно иметь доступ ко всем материалам, относящимся к аудиту и последующей работе по контракту.
- Работа по контракту, оказывающая критическое влияние на качество продукции, должна быть организована в соответствии с принципами анализа рисков (часть III настоящего стандарта). Уполномоченное лицо должно ознакомиться с результатами аудита, относящимися к критическому влиянию на качество продукции, до сертификации соответствующих серий.
- Следует проводить повторные аудиты согласно принципам анализа рисков.
3 Действия в случае непредвиденных отклонений
При соответствии активных субстанций, вспомогательных материалов, упаковочных материалов и лекарственных средств утвержденным спецификациям, уполномоченное лицо может подтвердить сертификацию и или сертифицировать серию, если произошли неожиданные отклонения в производственном процессе или аналитических методах контроля от требований регистрационного досье или требований настоящего стандарта (GMP). Отклонения должны быть тщательно расследованы и первопричина устранена. Это может потребовать внесения изменений в регистрационное досье для продолжающегося производства продукции.- Следует оценить влияние отклонений в соответствии с принципами анализа рисков (часть III настоящего стандарта). Анализ рисков должен включать следующее:
- Оценку возможного влияния отклонения на качество, безопасность и эффективность данной серии и заключение о том, что влияние отклонения незначительно.
- Рассмотрение необходимости включения данной серии в программу последующего контроля стабильности.
- В случае биологических лекарственных средств учет того, что любое отклонение от утвержденного процесса может оказать непредвиденное влияние на безопасность и эффективность.
4 Выпуск серии
- Выпуск серии лекарственного средства в реализацию допускается только после ее сертификации уполномоченным лицом согласно рассмотренному порядку. До завершения сертификации серия должна находиться на производственной площадке или быть отгружена с соблюдением условий карантина на другую площадку, которая утверждена для этой цели надзорным органом.
- Должны быть предусмотрены средства защиты от отправки несертифицированных серий на склад сбыта. Они могут быть физическими, например, разделение зон или нанесение этикеток, или электронными, например, с использованием аттестованных компьютерных систем. Пи перемещении несертифицированных серий с одной утвержденной площадки на другую должны сохраняться средства защиты от преждевременного выпуска.
- Этапы, необходимые для извещения площадки, на которую перемещается предназначенная для реализации продукция, о сертификации уполномоченным лицом, должны быть определены в техническом соглашении. Такое извещение уполномоченным лицом должно быть формальным и однозначным и быть выполнено согласно главе 4 части I настоящего стандарта (GMP).
5 Термины и определения
серия нерасфасованной готовой продукции (bulk production batch): Серия продукции с размером, установленным при регистрации лекарственного средства, готовая к фасовке в окончательную упаковку, либо находящаяся в индивидуальных упаковках и готовая для комплектования окончательных упаковок. Серия нерасфасованной продукции может содержать, например, жидкий нерасфасованный продукт, твердые формы (таблетки или капсулы) или наполненные ампулы. оценка соответствия серии готовой продукции (certification of the finished product batch): Документальное оформление соответствия серии готовой продукции установленным требованиям до выпуска серии в реализацию. подтверждение (confirmation): Подписанное свидетельство того, что процесс или испытания выполнены в соответствии с правилами GMP и требованиями, установленными при государственной регистрации, и согласованное в письменной форме с уполномоченным лицом, отвечающим за оценку соответствия серии готовой продукции до ее выпуска. серия готовой продукции (finished product batch): В контексте настоящего приложения означает серию продукции в окончательной упаковке, готовую к выпуску в реализацию. импортер (importer): Обладатель права (держатель лицензии) на импорт лекарственного средства. соглашение о взаимном признании (Mutual Recognition Agreement – MRA): Соглашение о вза- имном признании инспекций со страной, в которой производятся (из которой поставляются) ввозимые лекарственные средства. уполномоченное лицо (Qualified Person): См. пункт 50 раздела «Общие требования и определения».Приложение I
Содержание подтверждения об этапе производства лекарственного средства
[Головка бланка письма организации-производителя]- Наименование продукции и описание стадии производства (например, парацетамол 500 мг, таблетки, первичная упаковка – блистер)
- Номер серии
- Наименование и адрес площадки, где выполняется этап производства
- Ссылка на техническое соглашение по качеству (в соответствии с главой 7 части I настоящего стандарта)
- Заявление о подтверждении соответствия.
- Фамилия, имя и отчество уполномоченного лица, подтверждающего производство на данном этапе
- Подпись уполномоченного лица, подтверждающего производство на данном этапе
- Дата подписания
Основные принципы
В настоящем приложении приводятся основные принципы аттестации (испытаний) помещений, оборудования, вспомогательных систем и процессов производств лекарственных средств, которые могут использоваться в качестве дополнительных рекомендаций к активным субстанциям без дополнительных указаний в требованиях к их производству. Согласно требованиям настоящего стандарта производитель должен контролировать критические элементы производства путем проведения аттестации (испытаний) в течение жизненного цикла продукта и процесса. Следует документально оформлять любые запланированные изменения в помещениях оборудовании, вспомогательных системах и процессах, которые могут влиять на качество продукции и оценивать их влияние на соответствие требованиям (подтвержденным при аттестации) и стратегию (порядок) контроля. Следует также проводить аттестацию компьютерных систем, используемых в производстве лекарственных средств согласно приложению к настоящему стандарту. Рекомендуется учитывать соответствующие положения руководств ICH Q8, Q9, Q10 и Q11.Общие положения
В течение всего жизненного цикла лекарственного средства следует проводить анализ рисков. Объем работ по аттестации (испытаниям) следует определять исходя из оценки рисков для помещений, помещений, оборудования, вспомогательных систем и процессов. Ретроспективная аттестация более не рассматривается в качестве приемлемого метода. Могут использовать данные, относящиеся к аттестации (испытаниям), полученные из внешних источников за рамками программ производителя, при условии их обоснованности и уверенности в том, обеспечен достаточный контроль при получении таких данных.1 Организация и планирование работ по аттестации (испытаниям)
- Следует предусматривать необходимые работы по аттестации (испытаниям) в течение жизненного цикла помещений, оборудования, вспомогательных систем, процессов и продукции.
- Аттестация (испытания) должна проводиться только подготовленным персоналом, который работает по утвержденным методикам.
- Персонал, который выполняет аттестацию (испытания) должен оформлять протоколы о ее проведении согласно фармацевтической системе качества. Этот персонал необязательно должен входить в подразделения по контроль или обеспечению качества.
- Основные элементы аттестации (испытаний) должны быть четко определены программой аттестации (испытаний) или аналогичным документом.
- Эта программа или эквивалентный ей документ должны определять систем у аттестации и включать в себя следующие элементы (или давать ссылки на них):
- Цель проведения аттестации (политика аттестации);
- Организационную схема, включая функции и ответственность за проведении аттестации (испытаний);
- Перечень помещений, оборудования, вспомогательных систем и процессов, подлежащих аттестации;
- Порядок контроля изменений и отклонений при аттестации;
- Руководство по разработке допустимых критериев;
- Ссылки на существующие документы;
- Порядок проведения аттестации (испытаний), включая повторную аттестацию, если требуется.
- Для больших и сложных объектов планирование играет особую роль, и программа аттестации (испытаний) может быть разделена на несколько частей.
- При проведении аттестации (испытаний) следует выполнять анализ рисков. Следует проверять оценку рисков при получении новых данных и более глубокого понимания предмета после изменений или из опыта коммерческого производства, если требуется. Следует документально оформлять, как использовался анализ рисков при проведении аттестации (испытания).
- Для обеспечения полноты данных следует предусматривать необходимые проверки при проведении аттестации (испытаний).
2 Документация, включая программу аттестацию (испытаний)
- Правильное ведение документации играет важную роль для обеспечения информацией в течение всего жизненного цикла продукции.
- Все документы по аттестации (испытаниям) должны быть согласованы и утверждены уполномоченным на то персоналом в соответствии с системой качества.
- Следует четко определить взаимосвязь между документами в проведении аттестации (испытаний) сложных объектов.
- В протоколах о проведении аттестации (испытаний) следует четко обозначать критические системы, свойства и параметры с указанием допустимых пределов.
- Допускается объединять документы по аттестации, если это оправдано, например, протоколы аттестации в построенном и оснащенном состояниях.
- Если протоколы об аттестации и другая документация представлены третьей стороной, предоставляющей услуги по аттестации, то персонал производителя должен проверить эту документацию на соответствие внутреннему порядку производителя до ее приемки. Протоколы поставщиков могут быть дополнены документацией об испытаниях до начала эксплуатации.
- Любые существенные изменения к утвержденным протоколам в процессе эксплуатации (например, допустимых критериев, рабочих параметров и т. д.) следует оформлять документально и обосновывать.
- Результаты, не удостоверяющие установленным критериям, следует оформлять как отклонения и полностью расследовать в соответствии с установленным порядком. В протоколе аттестации следует рассмотреть любые относящиеся к ней факторы.
- Следует рассмотреть результаты аттестации и дать заключение об их соответствии заданным требованиям. Следует обосновать любые изменения и дать соответствующие рекомендации.
- Следует получить согласование ответственного лица на переход к следующему этапу аттестации (испытаний) либо в форме утверждения протокола аттестации, либо в форме отдельного итогового документа. При переходе к следующему этапу аттестации может быть дано условное согласование, если некоторые критерии не были полностью рассмотрены и если есть документальная оценка того, что отсутствует существенное влияние на следующий этап.
3 Этапы аттестации оборудования, помещений, вспомогательных систем и процессов
- При аттестации следует учесть все стадии от разработки задания на проектирование (технического задания) до завершения эксплуатации помещений, оборудования, вспомогательных систем и процессов. Ниже приведены основные стадии с перечислением входящих в них элементов, которые могут изменяться для конкретного объекта.
Задание на проектирование (техническое задание)
- В задании на проектирование (техническом задании) должны быть приведены требования к помещениям, оборудованию, вспомогательным системам и процессам. Следует предусмотреть выполнение существенных требований к качеству с учетом анализа рисков. Это документ должен служить критерием при аттестации на протяжении всего жизненного цикла.
Аттестация проекта (DQ)
- На этой стадии следует проверить соответствие проекта (конструкторской документации) помещений, оборудования, вспомогательных систем, процессов на соответствие требованиям GMP с документальным оформлением. Следует проверить соответствие заданию на проектирование (техническому заданию).
Приемосдаточные испытания на заводе-изготовителе (Factory Acceptance Testing – FAT)/ пусконаладочные испытания на месте эксплуатации (Site Acceptance Testing – SAT)
- Продавец может провести испытания оборудования, особенно нового и сложного до отгрузки.
- До начала монтажа следует проверить соответствие оборудования техническому заданию у поставщика, если требуется.
- В обоснованных случаях некоторые испытания могут быть проведены на заводеизготовителе(FAT) или в другом месте без проведения их на месте эксплуатации (SAT, стадии IQ/OQ), если может быть показано, что при транспортирование и монтаж не влияют на работоспособность оборудования.
- Приемо-сдаточные испытания (FAT) могут быть дополнены пуско-наладочными испытаниями (SAT) после приемки на месте эксплуатации.
Аттестация установленного оборудования, аттестация в построенном состоянии (IQ)
- Следует проводит аттестацию построенных (установленных) помещений, оборудования, вспомогательных и других систем.
- При аттестации установленного оборудования проверяются, как минимум:
- монтаж оборудования, трубопроводов, систем обслуживания и инструментов на соответствие чертежам и спецификации;
- проверка монтажа на соответствие документации;
- полнота и соответствие предоставляемой поставщиком документации по эксплуатации и техническому обслуживанию;
- наличие документации по калибровке (поверке);
- соответствие материалов установленным требованиям (проекту).
Аттестация в оснащенном состоянии (OQ)
- Аттестация в оснащенном состоянии (аттестация функционирующего оборудования) выполняется после успешного завершения аттестации установленного оборудования.
- При аттестации в оснащенном состоянии выполняются, как минимум:
- испытания и проверки, исходя из специфических особенностей процессов, систем и оборудования, на соответствие их заданным требованиям;
- испытания функционирования оборудования при рабочих параметрах, равных верхним и нижним допустимым пределам, т.е. в условиях «наихудшего случая».
- После завершения аттестации оборудования в оснащенном состоянии должны быть разработаны инструкции по эксплуатации и очистке, проведено обучение персонала и налажена система технического обслуживания.
Аттестация в эксплуатации (PQ)
- Аттестация в эксплуатации выполняется, как правило, после успешного завершения аттестации в построенном и оснащенном состояниях.
- При аттестации в эксплуатации выполняются, как минимум:
- испытания и проверки с использованием реальных материалов, аттестованных заменителей или имитаторов продукта при нормальных условиях эксплуатации для размеров серий, соответствующих наихудшему случаю. Следует обосновать периодичность отбора проб для подтверждения соответствия процесса заданным требованиям;
- испытания должны охватывать весь рабочий диапазон параметров процесса, если отсутствует документальное подтверждение этому диапазону на стадии разработки.
4 Повторная аттестация
- Следует проверять помещения, оборудование, вспомогательные и другие системы для подтверждения их соответствия заданным требованиям.
- При необходимости проведения повторной аттестации через определенные интервалы времени следует обосновать ее периодичность и задать критерии оценки. Следует также оценить возможность незначительных изменений с течением времени.
5 Аттестация (испытания) процесса
Общие положения
- Изложенные в настоящем разделе требования относятся к производству всех фармацевтических дозированных форм. Они включают первоначальную аттестацию новых процессов, последующую аттестацию модифицированных процессов, перенос процессов на площадке и текущий контроль процесса. Настоящее приложение исходит из того, что при разработке процесса заложена его устойчивость и это является основой успешной аттестации.
- Раздел 5 данного приложения следует использовать совместно с руководством Европейского агентства по лекарственным средствам (Guideline on process validation for finished products information and data to be provided in regulatory submissions – EMA/CHMP/CVMP/QWP/BWP/70278/2012, 12 November 2012).
- Назначением этого руководства является подготовка данных для представления в надзорные органы. В то же время требования GMP к аттестации процессов распространяются на все этапы жизненного цикла процесса.
- Этот подход применяется для установления связи продукции с разработкой процесса. Он обеспечивает проведение аттестации коммерческого процесса и поддержание процесса в соответствии с заданными требованиями в текущем производстве.
- Разработка процесса производства может проводиться на основе традиционного подхода или подхода с постоянными проверками. Но независимо от подхода следует показать устойчивость процесса и его способность обеспечивать качество продукции до ее выпуска в реализацию. При использовании традиционного подхода следует выполнять его перспективную аттестацию по соответствующей программе везде, где это возможно, до выдачи разрешения на выпуск продукции. Ретроспективная аттестация на является приемлемым подходом.
- При аттестации процесса производства новой продукции следует проверить процесс для всех значений эффективности продукции, предназначенной к выпуску и всех производственных площадок. Допускается применение метода исключений для новой продукции при наличии обоснования и хорошего знания стадии разработки процесса в сочетании с последующим контролем.
- В случае переноса процесса с одной площадки на другую или в пределах одной площадки число серий продукции для аттестации может быть снижено и использованием метода исключений. При этом нужно располагать знаниями о продукции, включая результаты прежней аттестации. При различных значениях эффективности, размерах серии и размерах упаковок (типах первичных упаковок) может применяться метод исключения (при наличии обоснования).
- При переносе существующей продукции на другую площадку следует обеспечить соответствие технологического процесса и его контроля регистрационному досье и относящимся к нему стандартам для данной продукции.
- При аттестации процесса следует установить, показатели качества и параметры процесса, имеющие важное значение для поддержания статуса «Аттестовано» и требуемого качества продукции, постоянно соответствуют заданным требованиям в ходе процесса. В документации следует четко указать, на основании чего параметры процесса или показатели качества отнесены к «критическим» или «некритическим», учитывая результата анализа рисков.
- Как правило, размер серии при аттестации должен быть равным размеру серии при промышленном выпуске продукции. Для других размеров серии следует дать обоснование согласно другим разделам настоящего стандарта.
- Следует провести аттестацию (испытания) оборудования, помещений, вспомогательных и других систем, участвующим в аттестации процесса. Методы испытаний следует аттестовать*.
- Для всей продукции основой для аттестации, независимо от используемого подхода, должна быть информация о процессе, содержащаяся в документах о ее разработке или в других источниках, если не указано иное.
- В производстве серий при аттестации процесса может участвовать персонал, занятый в его разработке, переносе на другую площадку или в выпуска продукции. К производству серий допускается только персонал, прошедший подготовку по правилам GMP, по утвержденной документации. Предполагается, что в производстве серий продукции при аттестации, занят то же персонал, что и в серийном производстве, что дает более полное понимание.
- Следует провести аттестацию (аудит) поставщиков критических исходных и упаковочных материалов до производства серий при аттестации процесса. В противном случае следует провести анализ рисков с документальным оформлением.
- Особое значение имеет знание процесса для обоснования параметров разработки процесса (если это применятся) и для разработки математических моделей (если они используются) для подтверждения порядка (стратегии) контроля.
- Если осуществляется выпуск в реализацию серий, произведенных при аттестации процесса, то это должно быть определено заранее. Условия производства таких серий должны полностью соответствовать требованиям GMP, включая критерии приемлемости при аттестации, контрольные параметры при постоянной проверке процесса (если предусмотрено) и соответствовать регистрационному досье и протоколу клинических исследований.
- Порядок аттестации процесса производства лекарственных средств для исследований приведен в приложении 15 к настоящему стандарту.
Текущая аттестация
- В исключительных случаях, когда есть острая потребность для пациента исходя из соотношения эффект/риск, допускается не завершать программу аттестации до начала серийного производства, прибегая к текущей аттестации. Решение о проведении текущей аттестации должно быть обосновано, включено в программу аттестации и утверждено лицами, имеющими на это право.
- В случае применения текущей аттестации следует получить достаточные данные для заключения об однородности серии продукции и ее соответствии заданным требованиям. Результат и заключение должны быть оформлены документально и быть доступными уполномоченному лицу при выдаче разрешения на реализацию серии.
- Не требуется аттестация методов испытаний, установленных стандартами и другими нормативными документами (прим. разработчика стандарта).
Традиционный процесс аттестации
- Размер серии при аттестации должен быть равным размеру серии при промышленном выпуске продукции для подтверждения воспроизводимости.
- Число серий и число проб при аттестации процесса следует определять исходя из анализа рисков, чтобы учесть изменения параметров в пределах допустимых значений, выявить тенденции и получить достаточные данные для оценки. Каждый производитель должен определить и обосновать число серий, чтобы показать высокий уровень уверенности в том, что процесс способен постоянно производить продукцию высокого качества.
- Для аттестации процесса считается достаточным выпустить три последовательных серии при обычных условиях (это не ставит под сомнение п. 5.19). Может быть обосновано другое число серий с учетом того, что используется стандартный метод производства и аналогичные продукты или процессы уже используются на площадке. Может потребоваться дополнение первоначальной аттестации на трех сериях другими данными, полученными при производстве последующих серий.
- Следует подготовить протокол аттестации процесса, в котором указываются критические параметры процесса, критические свойства для качества и допустимые критерии, которые должны быть основаны на данных о разработке процесса или документации о процессе.
- Протоколы аттестации процесса должны включать, как минимум, следующее:
- краткое описание процесса со ссылками на протоколы серий;
- обязанности и ответственность;
- перечень критических стадий процесса, которые подлежат исследованию;
- перечень критических параметров процесса с указанием допустимых пределов;
- перечень других (некритических) свойств и параметров, которые будут рассмотрены или будут результаты их контроля при аттестации с указанием причин, по которым они включены в перечень;
- перечень используемого при аттестации оборудования (в т. ч. контрольно-измерительные приборы и регистрирующее оборудование) с указанием наличия калибровки (поверки);
- перечень используемых аналитических методов (если требуется);
- предлагаемые виды внутрипроизводственного контроля и критерии приемлемости с указанием причин, почему они включены в состав этих работ;
- дополнительные испытания и критерии приемлемости;
- план отбора проб c обоснованием его;
- порядок оформления и оценки результатов;
- порядок выпуска серий и получения разрешения на их выпуск (если требуется).
Постоянный контроль процесса
- Для продукции, разработанной по принципу «качество за счет проектирования и разработки», когда в ходе разработки научно установлены стратегия контроля, которая обеспечивает высокую степень обеспечения качества, то может использоваться постоянный контроль процесса в качестве альтернативного подхода к традиционной аттестации процесса*.
- Следует определить метод контроля процесса. Следует разработать стратегию контроля на основе научного подхода для требуемых свойств исходных материалов, критических свойств для качества и критических параметров процесса для подтверждения выпуска продукции. Это должно также включать регулярную оценку стратегии контроля. В качестве методов может использоваться технология анализа процесса (Process Analytical Technology – PAT) и многовариантный статистический процесс контроля. Каждый производитель должен определить и обосновать число серий для демонстрации высокого уровня подтверждения того, что процесс постоянно производит продукцию заданного качества.
- Основные принципы п. п. 5.1 – 5.14 остаются в силе.
Гибридный подход
- Гибридный подход, т. е. сочетание традиционного подхода и постоянного контроля процесса может использоваться при наличии подробной информации о продукте и процессе и понимания, полученного из опыта производства и истории производства серий.
- Этот термин является надуманным, а сам подход неверным в принципе. Никакой разработки без уяснения задачи и понимания продукта (процесс) быть не может. Без пусконаладочных испытания (аттестации процесса) будет выпускаться продукция неизвестного качества, что будет установлено, возможно, только при серийном производстве (возможно, потому что объем и глубина текущего контроля всегда меньше, чем при аттестации). Подход по п. 5.23 не рекомендуется для применения в Российской Федерации. «Термин» приведен для сохранения целостности переводимого текста. См. также примечание АСИНКОМ к определению этого термина в разделе
- Этот подход может также использоваться при любых действиях по аттестации после изменений или по данным контроля выполняемого процесса, даже если проведена первоначальная аттестация согласно традиционному подходу*.
Контроль текущего процесса**
- П. п. 5.28-5.32 применимы ко всем трем подходам к аттестации процесса: традиционному, постоянному и гибридному.
- Производители должны контролировать качество продукции для подтверждения того, что процесс находится под контролем в течение всего жизненного цикла продукции. Требования могут быть модифицированы с учетом имеющегося понимания процесса и его выполнения.
- Следует регулярно рассматривать периодичность и объем текущего контроля процесса. В любой точке жизненного цикла продукции может потребовать изменение требований с учетом имеющегося понимания процесса и его выполнения.
- Постоянный контроль процесса следует проводить в соответствии с утвержденной инструкцией или эквивалентным документом и оформлять отчет о его результатах. Следует использовать статистические методы, где требуется, для подтверждения выводов с учетом изменчивости процесса и его способности соответствовать заданным требованиям.
- Следует применять постоянный контроль процесса в течение всего жизненного цикла продукции для подтверждения статуса «аттестовано» согласно Обзору качества продукции. Следует учитывать происходящие с течением времени изменения оценивать необходимость дополнительных мер, например, более частого отбора проб.
6 Контроль транспортирования
- Готовую лекарственные средства, лекарственные средства для исследований, нерасфасованную готовую продукцию (балк-продукт) и образцы следует транспортировать от производителя в условиях, установленных в регистрационном досье, с утвержденной этикеткой, спецификацией или в соответствии с документацией производителя.
- Транспортирование может представить опасность для качества из-за влияния изменяющихся факторов. Маршруты транспортирования должны быть четко определены. При контроле транспортирования следует учитывать сезонные и другие факторы.
- Для учета влияния изменяющихся факторов на процесс транспортирования следует проводить анализа рисков. Эти факторы отличаются от постоянно контролируемых параметров. К ним относятся, например, задержки при перевозке, отказ средств контроля, отбензинивание жидкого азота, чувствительности продукции другие факторы.
- Ввиду возможного влияния различных переменных факторов на процесс транспортирования следует организовать постоянных контроль и регистрацию любых критических условий окружающей среды, которые могут воздействовать на продукт, если не обосновано иное.
7 Аттестация процесса упаковывания
- Изменения параметров упаковочного оборудование может, особенно при первичной упаковке, могут оказать существенной влияние на целостность упаковки и выполнение ею своих функций, например, блистерной ленты, саше и стерильных компонентов. Поэтому следует проводить аттестацию оборудования для первичной и вторичной упаковки готовой и нерасфасованной продукции.
- Аттестацию оборудования для первичной упаковки следует проводить для максимальных и минимальных значений критических эксплуатационных параметров, например, температуры, скорости работы оборудования, давления сжатия и других факторов.
8 Аттестация вспомогательных систем
- Следует подтвердить качество пара, воды, воздуха, других газов и т. д. после завершения монтажа в соответствии с п. 3 данного приложения.
- Продолжительность и объем работ по аттестации должны учитывать сезонные колебания, если требуется, и назначение системы.
- Следует выполнить анализ рисков для случаев, когда возможен прямой контакт с продуктом, например, системы отопления, вентиляции и кондиционирования воздуха или непрямой контакт (теплообменники др.) с целью снижения риска неисправности
9 Аттестация методов контроля
- Все аналитические методы контроля, используемые при аттестации (в том числе процессов очистки), сами должны быть аттестованы с установлением пределов обнаружения и количественной оценки, при необходимости, в соответствии с главой 6 части I настоящего стандарта.
- При аттестации метода контроля микробиологической чистоты следует подтвердить отсутствие влияния продукта на результаты контроля (рост микроорганизмов).
- В случаях, когда требуется проводить контроль микробиологической чистоты поверхностей чистых помещений, следует выполнять аттестацию метода контроля для подтверждения того, что дезинфицирующие средства не влияют на результаты контроля (рост микроорганизмов).
10 Аттестация методов очистки
- Для подтверждения эффективности процесса очистки следует выполнять аттестацию методов очистки для всех поверхностей оборудования, вступающих в контакт с продуктом. Могут использоваться имитирующие средства для большей научной обоснованности аттестации. Если используется несколько видов однотипного оборудования, то следует обосновать выбор единицы оборудования, которая выбрана для аттестации.
- Важным критерием оценки эффективности процесса очистки является визуальный осмотр. Как правило, не допускается использование визуального осмотра в качестве единственного критерия для аттестации. Метод «проверять, пока не будет чисто» не заменяет аттестацию процесса очистки.
- Процесс аттестации очистки требует определенного времени и для некоторых видов продукции могут потребоваться аттестация и контроль после каждой серии, например, в случае лекарственных средств для исследований. Требуется получить достаточный объем данных для подтверждения того, что оборудование является чистым и пригодным к использованию.
- При проведении аттестации следует учесть степень автоматизации процесса очистки. При применении автоматических средств очистки следует аттестовать процесс при заданных (нормальных) значениях параметров вспомогательных систем и оборудования
- Для всех процессов очистки следует определить переменные величины, которые влияют на эффективность очистки и эксплуатацию, например, влияние операторов, степень подробности инструкций, например длительность ополаскивания и т. д. Если это переменные не определены, то следует применить метод наихудшего случая в качестве основы для анализа процесса очистки.
- Пределы переноса остатков продукта на другой продукт следует определять на основе токсикологической оценки (см. Раздел 6 Части III настоящего стандарта). Обоснование этих пределов следует оформлять документально с учетом анализа рисков, который включает в себя все вспомогательные факторы. Следует установить пределы для удаления всех используемых моющих (дезинфицирующих) средств. Допустимые пределы должны учитывать возможный кумулятивный (суммарный) эффект нескольких видов оборудования в технологической линии.
- Терапевтические макромолекулы и пептиды изменяют свои свойства и деградируют под воздействием экстремальных значений рН и/или тепла и могут потерять фармакологическую активность. В этих случаях токсикологическая оценка не применяется.
- Если не удается контролировать специфические остатки продукции, то могут быть выбраны другие параметры, например, общий органический углерод и проводимость.
- При проведении аттестации процесса очистки следует учитывать риск загрязнения микроорганизмами и эндотоксинами.
- Следует учитывать влияние длительности интервалов времени между окончанием процесса и очисткой, а также очисткой и началом следующего процесса.
- При производстве циклами следует учитывать удобство поведения очистки после завершения цикла и определить максимально допустимую величину цикла (продолжительность и/или число серий в цикле).
- Ели при аттестации процесса очистки используется метод наихудшего случая, то следует дать научное обоснование выбору наихудшего продукта и оценить влияние новых продуктов на производство. Критерии для выбора наихудшего случая могут включать в себя растворимость, пригодность к очистке, токсичность и активность.
- В методике аттестации процесса очистки должны быть указаны точки отбора проб, обоснование для выбора точек и допустимые пределы и даны ссылки на них.
- Отбор проб следует выполнять методами мазков и/или смывов или другим способом в зависимости от технологического оборудования. Метод и материалы для отбора проб не должны влиять на результат. Для всех материалов, вступающих в контакт с продуктом, должна быть показана эффективность всех используемых методов отбора проб.
- Число раз (повторений) процесса очистки при проведении аттестации следует определять на основе анализа рисков, при этом каждый раз должно выполняться соответствие принятым критериям.
- Если процесс очистки неэффективен или не применим для некоторых видов оборудования, то следует предусматривать специализированное оборудование или принять другие меры для каждого продукта согласно главам 3 и 5 Части I настоящего стандарта.
- При выполнении очистки ручным методом подтверждение эффективности очистки становится особенно важным, при этом следует установить эффективность очистки.
11 Контроль изменений
- Контроль изменений является важной частью обеспечения информацией и должен выполняться в рамках фармацевтической системы качества.
- Внесение изменений в исходные материалы, компоненты продукции, процессы, оборудование, помещения номенклатуру продукции, методы производства и контроля, размеры серий, параметры для разработки или другие изменения в течение жизненного цикла, которые могут повлиять на качество продукта или воспроизводимость процесса, должно выполняться по специально разработанным инструкциям.
- Следует учесть влияние изменений на параметры для разработки (где они присутствуют) относительно заданных параметров в регистрационном досье и оценить необходимость в извещении надзорных органов.
- Для оценки намечаемых изменений следует проводить анализ рисков в плане возможного влияния изменений на качество продукции, фармацевтическую систему качества, документацию, аттестацию, официальный статус (регистрацию и др.), калибровку, техническое обслуживание и другие системы во избежание непредусмотренных последствий и с целью планирования аттестации процесса, проверок и повторной аттестации.
- Изменения следует согласовать и утвердить ответственными лицами или уполномоченным на то персоналом в соответствии с фармацевтической системой качества.
- Следует рассматривать вспомогательные материалы (копии документов и др.) и устанавливать их влияние до утверждения.
- Следует оценивать эффективность внесения изменений для оценки их полезности, где требуется.
Термины и определения
Приведенные термины, относящиеся к аттестации и испытаниям, не включены в раздел «Термины и определения» настоящего стандарта, но используются в данном приложении. Метод исключения (bracketing approach): метод аттестации, основанный на научном подходе и анализе рисков, согласно которому при аттестации проверяются только серии с экстремальными (наихудшими) параметрами, например, эффективность, размер серии и/иди размер упаковки. Предполагается, что аттестация для экстремальных значений параметров действительна и для промежуточных значений. В случае необходимости аттестовать процесс для различных значений эффективности допускается применять метод исключения, если эффективности одинаковы или сильно зависят от состава, например, для таблеток с различным весом после прессования при одном базовом процессе гранулирования или разных капсул, наполнение которых осуществлено при разных весах из одного базового состава в капсулы различных размеров. Аттестация процесса очистки (cleaning validation): Документальное подтверждение того, что утвержденный метод очистки неизменно удаляет остатки предыдущего продукта или моющих средств с оборудования, так, что они находятся ниже предельно допустимых значений переноса на другой продукт. Проверка эффективности очистки (cleaning verification): Проверка путем проведения химического анализа, что после каждого производства каждой серии (цикла производства) остатки предыдущего продукта или моющих средств на оборудования находятся ниже предельно допустимых значений переноса на другой продукт. Анализ рисков (quality risk management): системный процесс оценки, контроля и рассмотрения рисков для качества на протяжении жизненного цикла. Оценка риска (Risk assessment): Систематизированный процесс сбора информации для принятия решения в ходе анализа риска. Он включает в себя установление опасностей, анализ и оценку рисков, связанных этими опасностями. – это из ICH Q9 Аттестация в эксплуатации (performance qualification; PQ): Документальное подтверждение того, что системы и оборудование работают эффективно и с воспроизводимыми показателями в соответствии с промышленным регламентом, технологическими инструкциями и спецификацией на продукт. Аттестация проекта (design qualification; DQ): Документальное подтверждение того, что проект производства (в т. ч. помещения, системы и оборудование) соответствуют своему назначению*. * То есть. выполнен в соответствии с заданием на проектирование, настоящим стандартом и другими Постоянный контроль процесса (continuous process verification): метод, который в отличие от аттестации процесса, предусматривает непрерывный контроль и оценку функционирующего процесса*. Стратегия контроля, порядок контроля (Control Strategy): Плановый комплекс мер по контролю, построенный исходя из понимания продукта и процесса в текущее время, обеспечивающий выполнение требований к процессу и качеству продукции. Эти меры могут включать контроль параметров и свойств, относящихся к субстанциям лекарственных средств, материалов и компонентов лекарственных средств, условий работы помещений и оборудования, внутрипроизводственный контроль, спецификации на готовую продукцию и связанные с ними методы и периодичность контроля (ICH Q10). Критический параметр процесса (critical process parameter – CPP): Параметр процесса, изменения которого влияют на критические свойства для качества и который, в связи с этим, подлежит контролю или регулированию обеспечения выпуска продукции заданного качества (ICH Q8)/ Критическое свойство для качества (quality attribute – CQA): Физическое, химическое, биологическое или микробиологическое свойство или параметр, которые должны поддерживаться в заданных пределах, диапазоне или распределении для обеспечения выпуска продукции заданного качества (ICH Q8). Параметры для разработки (Design Space): Многомерная комбинация и взаимодействие переменных (например, свойств материалов) и параметров процесса, которые обеспечивают выполнение требований к качеству (ICH Q8). Обеспечение информацией (Knowledge Management): Систематический подход к получению, анализу, хранению и распространению информации о продукции, технологических процессах и компонентах (ICH Q10). Жизненный цикл (cycle): Все этапы жизни продукции, оборудования или производств от начала разработки и использования до прекращения выпуска. Аттестация процесса (process validation): Документальное подтверждение того, что процесс, выполняемый в рамках установленных параметров, протекает эффективно и с воспроизводимыми параметрами, производя лекарственное средство, удовлетворяющее всем заданным требованиям к продукции и ее качеству. Аттестация установленного оборудования, аттестация в построенном состоянии (installation qualification; IQ): Документальное подтверждение того, что монтаж помещений, систем и оборудования (установленных или измененных) выполнен в соответствии с проектом и другой технической документацией. Аттестация в оснащенном состоянии (operational qualification; OQ): Документальное подтверждение того, что помещения, системы и оборудование (установленные или измененные) функционирует в соответствии с предъявляемыми требованиями во всех режимах работы. Получение продукта (Product Realisation): Получение продукта с показателями качества, соответствующими нуждам пациентов, профессионалов здравоохранения и надзорных органов (включая соответствие лицензии) и внутренним требованиям заказчиков (ICH Q10). Качество за счет проектирования или разработки (quality by design): систематический подход, который начинается с уяснения задачи с акцентом на понимание продукта и процесса и управления процессом на основе научного подхода и анализа рисков**. Имитирующий материал (simulated agent): Материал, который по своим физическим и, по возможности, химическим характеристикам (например, вязкости, размерам частиц, рН и пр.) близок продукту, к которому относится аттестация. Контроль изменений (change control): Документально оформленный порядок, согласно которому представители различных специальностей рассматривают предложенные или существующие изменения, которые могут повлиять на статус «Аттестовано» для помещений, оборудования или процессов и определяют необходимость действий, которые должны обеспечить и документально оформить поддержание системы в статусе «Аттестовано». Наихудший случай (worst case): Условия или комплекс условий, относящихся к верхним и нижним предельным значениям параметров производственного процесса и связанных с ними факторов, определенных документацией, которые могут привести к самой высокой вероятности появления брака в процессе или продукте по сравнению с идеальными условиями. Такие условия не всегда обязательно приводят к браку в процессе или продукте. Перспективная аттестация (prospective validation): Аттестация, выполняемая до начала серийного производства продукции, предназначенной для реализации. нормативными документами (прим. разработчика стандарта). * Оригинал правил GMP EC содержит также термин «Ongoing Process Verification», определяемый как «документальное подтверждение соответствия коммерческого процесса заданным требования» и трактуемый как синоним термина «continuous process verification» (прим. разработчика стандарта). ** Этот термин является надуманным. Никакой разработки без уяснения задачи и понимания продукта (процесса) быть не может, в рамках нормальной логики, которая была всегда. «Термин» приведен в данном тексте для сохранения целостности переводимого текста (прим. разработчика стандарта). Состояние контроля (state of control): Условие, при котором комплекс мер по контролю непрерывно подтверждает соответствие процесса и качества продукции заданным значениям. Традиционный подход (traditional approach): подход к разработке продукции, при котором заданы требования и условия эксплуатации для обеспечения воспроизводимости. Задание на проектирование, техническое задание на разработку (User Requirements Specification – URS): Комплекс необходимых и достаточных требований пользователя, собственника или разработчика для разработки проекта (конструкторской документации исходя из назначения объекта или оборудования*.Общие положения
Лекарственные средства для клинических исследований должны быть изготовлены в соответствии с правилами производства лекарственных средств (GMP), изложенными в настоящем стандарте. Следует принимать во внимание и другие руководства с учетом стадии разработки препарата. Методики работы должны быть гибкими и допускать внесение изменений по мере получения дополнительных данных о процессе и соответствовать стадии разработки препарата. При клинических исследованиях может возникнуть дополнительный риск для субъектов исследований по сравнению с пациентами, которые принимают имеющиеся на рынке препараты. Целью правил GMP в применении к производству лекарственных средств для клинических исследований является исключение риска для субъектов исследований и влияния на результаты клинических исследований недостаточных безопасности, качества или эффективности, обусловленных недостатками в производстве. В равной мере эти требования предназначены для обеспечения постоянства от серии к серии одного и того же лекарственного средства для исследований, используемого в одном или в разных клинических исследованиях, а также документального оформления и обоснования изменений в процессе разработки исследуемого лекарственного средства. Производство лекарственных средств для исследований связано с дополнительной сложностью по сравнению с зарегистрированными лекарственными средствами из-за отсутствия установленного порядка, разных схем клинических исследований и, как следствие, разных требований к упаковкам, необходимости рандомизации и кодирования (маскирования, использования слепого метода), а также из-за большого риска перекрестных загрязнений и перепутывания препаратов. Более того, данные об эффективности и токсичности препарата могут быть неполными, процесс может быть не аттестован в полном объеме или могут использоваться зарегистрированные лекарственные средства, которые были переупакованы или модифицированы. В связи с этим персонал должен в полной мере понимать правила GMP в отношении лекарственных средств для исследований и пройти соответствующую подготовку. Должно быть установлено взаимодействие со спонсорами клинических исследований, на которых лежит вся ответственность по вопросам клинических исследований, включая качество лекарственных средств для исследований. Возросшая сложность технологических процессов требует применения высокоэффективной системы обеспечения качества. В данном приложении также установлены требования к оформлению заказов, отгрузке, транспортированию и возврату материалов для клинических исследований, которые взаимосвязаны и дополняют руководство по клиническим исследованиям (Good Clinical Practice). Примечания- Лекарственные средства, не предназначенные для исследования
- Лицензирование производства и подготовка к применению
- растворение или диспергирование лекарственного средства для исследований для его введения пациенту, или
- разведение или смешивание лекарственных средств для исследований с другим веществом (веществами), используемого(ых) как носитель с целью введения лекарственного средства.
Термины и определения
слепой метод, кодирование, маскирование (blinding): Процедура, при которой одна или более сторон, участвующих в исследовании, остаются в неведении относительно цели(ей) данного исследования. Единичный слепой метод означает неосведомленность субъектов исследования, а двойной слепой метод – неосведомленность о целях клинического исследования субъектов исследования, исследователей, наблюдателей и, в некоторых случаях, лиц, анализирующих полученные данные. В отношении лекарственного средства для клинических исследований слепой метод означает преднамеренное маскирование идентичности этого лекарственного средства в соответствии с указаниями спонсора. Декодирование (снятие маскировки) означает раскрытие информации об идентичности препарата. клиническое исследование (clinical trial): Исследование, проводимое с участием человека в качестве субъекта для выявления или подтверждения клинических, фармакологических и/или других фармакодинамических эффектов исследуемого лекарственного средства, и/или нежелательных реакций на него, и/или изучения его всасывания, распределения, метаболизма и выведения с целью оценки его безопасности и/или эффективности. препарат сравнения (comparator product): Разрабатываемое или имеющееся на рынке лекарственное средство (для положительного контроля) или плацебо, используемое для целей сравнения при проведении клинических исследований. лекарственное средство для исследований (investigational product): Дозированная лекарственная форма активной фармацевтической субстанции или плацебо, исследуемая или используемая в качестве препарата сравнения при проведении клинического исследования, включая уже зарегистрированные лекарственные средства, если способ их применения или производства (лекарственная форма или упаковка) отличается от зарегистрированной формы, или в случае их использования по еще не одобренным показаниям, или для получения дополнительной информации об уже зарегистрированной дозированной лекарственной форме. исследователь (investigator): Лицо, ответственное за проведение клинического исследования в месте его выполнения. При проведении клинического исследования группой лиц исследователем является руководитель группы, который может называться ведущим исследователем. производитель/импортер лекарственного средства для клинических исследований (manufacturer/importer of investigational medicinal products): Лицо, имеющее право на производство (импорт) исследуемого лекарственного средства, оформленное в установленном порядке. заказ (order): Задание на приготовление, упаковку и/или доставку определенного количества единиц лекарственного средства для клинических исследований. досье на лекарственное средство (product specification file): Комплект документов, содержащих всю информацию, необходимую для составления подробных инструкций по изготовлению, упаковке, контролю качества, выдаче разрешения на реализацию серии и отгрузке лекарственного средства для клинических исследований. рандомизация (randomization): Присвоение субъектам исследований или контрольным группам обозначений, содержащих элемент случайности, с целью снижения возможности необъективного заключения. код рандомизации (randomization code): Перечень, содержащий описание лечения каждого субъекта исследований с учетом рандомизации. отгрузка (shipping/dispatch): Операции по упаковке и транспортированию заказанных лекарственных средств для клинических исследований. спонсор (sponsor): Физическое или юридическое лицо, являющееся инициатором клинического исследования и несущее ответственность за его организацию и финансирование.Обеспечение качества
- Система обеспечения качества, разработанная и проверенная производителем или импортером, должна соответствовать требованиям настоящего стандарта, относящимся к лекарственным средствам для исследований, должна быть документально оформлена и быть доступна спонсору клинического исследования.
- Спецификации и технологические инструкции на лекарственные средства для клинических исследований могут изменяться в процессе их разработки. В связи с этим должен быть организован их контроль и прослеживаемость всех изменений.
Персонал
- Весь персонал, имеющий отношение к лекарственным средствам для клинических исследований, должен пройти подготовку, связанную со спецификой данного вида продукции.
- Уполномоченное лицо несет особую ответственность за то, чтобы выполнялись все требования GMP. Для этого оно должно иметь хорошую подготовку в области разработки препаратов, процессов и клинических исследований. Руководство для уполномоченного лица по оценке (сертификации) лекарственных средств для клинических исследований приведено в пунктах 38 – 41 настоящего приложения.
Помещения и оборудование
- При работе с лекарственными средствами для исследований информация о токсичности, эффективности и сенсибилизирующей способности может быть неполной, в связи с чем следует уделить особое внимание сведению к минимуму риска перекрестных загрязнений. Конструкция оборудования и проект помещений, методы испытаний и контроля и допустимые концентрации остатков после очистки должны учитывать природу этих рисков. Следует обратить внимание на организацию работы циклами (кампаниями), если это целесообразно. При выборе моющего средства следует учесть растворимость в нем препарата.
Документация
Спецификации и инструкции
- Спецификации (на исходные материалы, первичные упаковочные материалы, промежуточные продукты, нерасфасованную и готовую продукцию), промышленные регламенты, технологические инструкции, а также инструкции по упаковке продукции должны быть, по возможности, полными для существующего уровня знаний. По мере разработки препарата их следует периодически пересматривать (при необходимости). Каждая новая версия документа должна учитывать последние данные, используемую технологию, нормативные и фармакопейные требования со ссылкой на предыдущую версию для обеспечения прослеживаемости. Любые изменения, которые могут иметь последствия для качества препарата, в частности для его стабильности и биоэквивалентности, следует вносить в соответствии с письменными требованиями.
- Любые изменения следует оформлять документально. Последствия для качества препарата и на последующие клинические испытания необходимо анализировать и оформлять документально.
Заказ
- Заказ должен содержать требование на изготовление и/или упаковку определенного числа единиц продукции и/или ее отгрузку. Заказ производителю дается спонсором или лицом, действующим по его поручению. Заказ должен быть оформлен в письменном виде (но может передаваться и электронным способом) и быть достаточно четким во избежание разночтений. Заказ должен быть официально утвержден и иметь ссылку на утвержденное досье на лекарственное средство или протокол клинических испытаний.
Досье на лекарственное средство
- Досье на лекарственное средство должно непрерывно обновляться по мере разработки препарата, обеспечивая логическую связь с предыдущими версиями. Досье должно включать в себя следующие документы (или содержать на них ссылки):
- спецификации и аналитические методики для исходных и упаковочных материалов, промежуточной, нерасфасованной и готовой продукции;
- технологические процессы;
- методики внутрипроизводственного контроля;
- утвержденную копию этикетки;
- протоколы клинических исследований и коды рандомизации (при необходимости);
- технические соглашения с заказчиками по контрактам (при необходимости);
- данные о стабильности;
- условия хранения и транспортирования.
Промышленные регламенты и технологические инструкции
- Каждая производственная операция или операция по отгрузке должна выполняться в соответствие с четкой и достаточно полной письменной инструкцией и сопровождаться оформлением протокола. Если операция не является повторяемой, то не обязательно составлять промышленный регламент и технологические инструкции. Протоколы имеют особое значение для подготовки окончательных текстов документов, которые будут использоваться при серийном производстве после получения регистрационного удостоверения.
- Информацию, содержащуюся в досье, следует использовать при разработке подробных инструкций по изготовлению, упаковке, испытаниям для контроля качества, условиям хранения и транспортирования.
Инструкции по упаковке
- Лекарственные средства для исследований должны упаковываться индивидуально для каждого субъекта испытаний. Количество единиц упаковываемой продукции должно быть определено до начала операций по упаковке с учетом количества единиц, необходимых для проведения контроля качества, и архивных образцов. После окончания упаковки и маркировки необходимо подвести баланс (выполнить сопоставление) каждого вида упаковочных материалов, нерасфасованной и готовой продукции для каждой стадии производства.
Протоколы производства, испытаний и упаковки серии продукции
- Протоколы производства и упаковки серии продукции должны содержать подробную информацию, достаточную для прослеживания всей последовательности операций, а также все существенные замечания, дающие новую информацию о продукте и внесению изменений, позволяющих получить дополнительные данные о продукции, усовершенствовать производственный процесс и технологические инструкции.
- Протоколы производства серий продукции должны храниться не менее пяти лет после завершения или официального прекращения последнего клинического испытания, в котором была использована эта серия*.
Производство
Упаковочные материалы
- В спецификациях и инструкциях (методиках) контроля качества должны быть указаны меры по предотвращению случайного снятия кодирования (маскировки) из-за различий внешнего вида разных серий упаковочных материалов.
Технологические операции
- На стадии разработки лекарственного средства следует определить критические параметры и виды внутрипроизводственного контроля для контроля технологического процесса. Временные параметры и виды внутрипроизводственного контроля могут быть получены из прошлого опыта, в том числе, из предыдущих исследований. Руководящий персонал должен уделять особое внимание разработке инструкций и постоянному их развитию с учетом опыта, приобретаемого в процессе производства. Контролируемые параметры должны быть обоснованы в соответствии с имеющейся в данное время информацией.
- Не обязательно проводить аттестацию (испытания) технологических процессов производства лекарственных средств для исследований в объеме, предусматриваемом для серийного производства, но помещения и оборудование должны быть аттестованы. Для стерильных лекарственных средств аттестация (испытания) процессов стерилизации должна проводиться по тем же стандартам, что и для серийного производства продукции, разрешенной к реализации.
- Аттестация (испытания) асептических процессов представляет особую трудность при малых размерах серий продукции. В этих случаях число единиц, наполняемых средами, может быть равно наибольшему размеру серии продукции. По возможности (в том числе для имитации процесса) следует наполнять средами большее число единиц продукции для обеспечения большей достоверности результатов. Наполнение и герметизация являются, преимущественно, ручными или полуавтоматическими операциями, представляющими риск для стерильности. В связи с этим следует уделить повышенное внимание обучению персонала и аттестации методов асептического производства для конкретных операторов.
- Эти требования установлены в ст. 9 Директивы 2003/94/ЕС.
Требования к препарату сравнения
- При существенных изменениях лекарственного средства объем информации о нем (например, по стабильности, сравнительной растворимости, биодоступности) должен быть доступным для демонстрации того, что эти изменения не окажут существенного влияния на исходные параметры качества этого лекарственного средства.
- Срок годности препарата сравнения, указанный на первоначальной упаковке, не может быть таким же для переупакованного в другую упаковку препарата, поскольку при этом может быть не обеспечен эквивалентный уровень защиты или нарушена совместимость с продуктом. Поэтому спонсор (или лицо, действующее от его имени) обязан определить соответствующий срок годности (с учетом вида препарата, характеристик упаковки и условий хранения). Новый срок годности должен быть обоснован и не может превышать указанный на первоначальной упаковке. Этот срок должен быть совместим со сроком хранения и длительностью клинических исследований.
Операции по кодированию (маскированию)
- При применении кодирования (маскирования) должна существовать система, гарантирующая маскировку и ее поддерживание. В то же время эта система должна позволять идентифицировать, при необходимости, кодированный («слепой») препарат, в том числе номера серий до маскирования. Следует предусмотреть возможность быстрого снятия маскировки в экстренных случаях.
Код рандомизации
- В инструкциях должны быть четко описаны все операции (формирование, распространение, обращение и хранение) с любым кодом рандомизации, используемым для упакованных лекарственных средств для клинических исследований, а также методы снятия кода. Следует ввести соответствующую документацию.
Операции по упаковке
- При упаковывании лекарственного средства для исследований может оказаться необходимым одновременное обращение с различными продуктами на одной и той же упаковочной линии. Риск перепутывания продуктов должен быть сведен к минимуму за счет применения специальных методов работы и/или специализированного оборудования и обучения персонала.
- Операции по упаковке и маркировке лекарственных средств для исследований могут быть более сложными и подверженными ошибкам (которые труднее выявлять), чем при производстве для реализации, особенно при кодировании продуктов с похожим внешним видом. В связи с этим требуется принимать особые меры по предотвращению ошибок в маркировке, например, за счет ведения баланса (сопоставления) этикеток, очистки линии, внутрипроизводственного контроля специально подготовленным персоналом.
- Упаковка должна гарантировать сохранность лекарственного средства в требуемом состоянии при транспортировании и хранении в промежуточных пунктах. Вторичная упаковка должна быть такой, чтобы было сразу заметно любое нарушение ее целостности при транспортировании.
Маркировка
- В таблице приведены требования, содержащиеся в пунктах 26 – 30 настоящего приложения. Маркировка должна обеспечивать защиту субъекта исследований, возможность прослеживания и идентификации препарата и испытания и способствовать правильному применению лекарственного средства для исследований*. Маркировка должна содержать следующую информацию, если не обосновано ее отсутствие (например, при наличии централизованной электронной системы рандомизированного кодирования):
- b) лекарственную форму, способ введения, количество дозированных единиц, и в случае проведения открытого исследования – наименование/шифр лекарственного средства и его активность/эффективность;
- Эти требования установлены ст. 15 Директивы 2003/94/ЕС.
- d) номер (код) исследования, позволяющий идентифицировать исследование, медицинское учреждение, исследователя и спонсора, если это не указано в другом месте;
- фамилию и инициалы исследователя (если не указано в перечислениях а) или d));
- инструкции по применению (может быть приведена ссылка на листок-вкладыш, либо другой пояснительный документ, предназначенный для субъекта клинического исследования или лица, которое вводит препарат);
- Надпись «Только для клинических исследований» или аналогичная формулировка;
- условия хранения;
- срок использования с указанием месяца и года таким образом, чтобы избежать любой неопределенности (может быть указана дата, до которой необходимо использовать препарат, срок годности или дата повторного контроля);
- надпись «Хранить в недоступном для детей месте» за исключением случаев, когда препарат предназначен для использования только в условиях стационара.
- Адрес и номер телефона основного контактного лица для передачи информации относительно препарата, клинического исследования и срочного декодирования могут быть не указаны на этикетке, если субъекту исследования предоставлены листок-вкладыш или карточка, на которой указаны эти данные, а также даны инструкция держать их при себе постоянно.
- Данные должны быть приведены на официальном языке (языках) страны; где будет применяться лекарственное средство для исследований. Данные, приведенные в пункте 26 данного приложения, должны находиться как на первичной, так и на вторичной упаковке (данные могут быть не указаны на первичной упаковке в случаях, указанных в пунктах 29 и 30 настоящего приложения). Требования к информации, приводимой на первичной и вторичной упаковках, приведены в таблице 13.1. Также на этикетках может быть приведена информация на других языках.
- Если препарат подготовлен для субъекта исследований или лица, которое вводит препарат, то на первичной упаковке вместе со вторичной упаковкой, которые следует оставлять вместе, и на вторичной упаковке должны содержаться данные, приведенные в пункте 26 настоящего приложения. На этикетке первичной упаковки (или любого укупоренного дозирующего устройства, содержащего первичную упаковку) необходимо указать следующую информацию:
- b) лекарственную форму, способ введения (можно не указывать для твердых лекарственных форм для орального применения), количество дозированных единиц и в случае проведения открытого исследования, наименование/шифр лекарственного средства, а также активность/эффективность;
- d) номер (код) исследования, позволяющие идентифицировать исследование, медицинское учреждение, исследователя и спонсора, если все это не указано в другом месте;
- Если первичной упаковкой является блистер или она имеет малый размер, например ампулы, на которых не могут быть размещены данные, приведенные в пункте 26 настоящего приложения, должна быть предусмотрена вторичная упаковка с этикеткой, содержащей эти данные. Однако на первичной упаковке должны быть указаны:
- b) способ введения (можно не указывать для твердых лекарственных форм для орального применения) и в случае проведения открытых исследований, наименование/шифр лекарственного средства, а также активность/эффективность;
- d) номер (код) исследования, позволяющий идентифицировать исследование, медицинское учреждение, исследователя и спонсора, если все это не указано в другом месте;
- Для пояснения указанной выше информации могут быть использованы символы или пиктограммы. Может быть представлена дополнительная информация, предостережение и/или инструкции по обращению с препаратом.
- В некоторых случаях* при проведении клинических исследований на первичной упаковке дополнительно должны быть приведены следующие данные (так, чтобы они не закрывали первоначальную этикетку):
- – наименование (имя) спонсора, исследовательского учреждения или исследователя, с которым заключен контракт;
- – номер (код) исследования, позволяющий идентифицировать медицинское учреждение, исследователя и субъекта исследований.
- Если необходимо изменить дату использования лекарственного средства, следует нанести на упаковку дополнительную этикетку. На дополнительной этикетке должна быть указана новая дата, до которой следует использовать препарат, а также повторно указан номер серии. Дополнительную этикетку можно наклеивать поверх старой даты использования, но она не должна закрывать исходный номер серии, что связано с контролем качества. Эту операцию можно осуществлять на производственном участке, который имеет на это право. При необходимости это может осуществляться в исследовательском учреждении фармацевтом, проводящим клинические испытания либо под его наблюдением, а также другим медицинским работником в соответствии с требованиями действующего законодательства. Если это невозможно, операцию может осуществлять лицо, которое прошло соответствующее обучение. Операцию следует осуществлять согласно принципам GМР в соответствии со специальными и стандартными методиками и, при необходимости, по контракту; операцию должно контролировать второе лицо. Дополнительное этикетирование следует тщательным образом документировать как в документах клинических исследований, так и в протоколах серии.
Контроль качества
- Поскольку процессы могут быть не стандартными или не в полной мере аттестованными, возрастает значение испытаний для обеспечения гарантии того, что каждая серия продукции соответствует спецификации на нее.
- Контроль качества необходимо осуществлять в соответствии с досье на лекарственное средство и согласно информации, предоставленной спонсором компетентному органу при запросе разрешения на проведение клинических исследований**. Следует проводить проверку эффективности кодирования и результаты ее оформлять документально.
- Необходимо хранить образцы для достижения двух целей: во-первых, для проведения аналитических исследований, во-вторых, чтобы иметь образец готового продукта. В связи с этим образцы могут быть разделены на две группы:
- Место хранения контрольных и архивных образцов должно быть определено в соглашении между спонсором и производителем(ями). К таким местам необходимо обеспечить своевременный доступ представителей надзорных органов.
- Эти случаи указаны в статье 14 Директивы 2001/20/ЕС и относятся к ситуациям, когда:
- нет необходимости в раздельных процессах производства или упаковки;
- при исследовании используются лекарственные средства, зарегистрированные, произведенные или импортированные в соответствии с действующим законодательством;
- в исследованиях принимают участие пациенты с теми заболеваниями, которые соответствуют показаниям к применению, указанным в инструкции по применению, утвержденной при регистрации лекарственного средства.
Выдача разрешения на выпуск серий
- Не допускается выдача разрешения на выпуск лекарственных средств для исследований (см. пункт 43 настоящего приложения) до тех пор, пока уполномоченное лицо не удостоверит выполнение установленных требований и требований данного приложения* (см. пункт 39 настоящего приложения). Уполномоченное лицо должно учитывать факторы, приведенные в пункте 40 настоящего приложения.
- На выполнение уполномоченным лицом своих обязанностей в отношении лекарственных средств для исследований влияют разные факторы.
- препарат произведен в Российской Федерации, но не зарегистрирован в Российской Федерации: необходимо указать, что лекарственные средства для исследований произведены и проверены в соответствии с требованиями GMP, изложенными в настоящем стандарте, досье на лекарственное средство и информацией, предоставленной спонсором компетентному органу при запросе разрешения на проведение клинических исследований**;
- препарат находится на рынке Российской Федерации, поставляется производителем или дистрибьютором, который имеет на то право, и зарегистрирован в Российской Федерации независимо от того, где производится препарат. Его обязанности указаны выше, но объем представленных данных может быть ограничен подтверждением того, что препараты соответствуют уведомлению/запросу на разрешение проведения клинических исследований и любой последующей обработки с целью кодирования, специальной упаковки или маркировки для этого исследования. Досье на лекарственное средство также может быть ограниченным по объему (см. пункт 9 данного приложения);
- препарат ввезен из другой страны: необходимо указать, что он произведен и проверен в соответствии с требованиями GMP (как минимум, эквивалентными изложенным в настоящем стандарте), досье на лекарственное средство и информацией, предоставленной спонсором компетентному органу при запросе разрешения на проведение клинического исследования***. Если лекарственные средства для исследований ввезены из другой страны и являются объектом соглашения, принятого между Российской Федерацией и этой страной, например таким, как соглашение о взаимном признании, любое подобное соглашение предусматривает применение эквивалентных стандартов GMP в отношении этого препарата. При отсутствии соглашения о взаимном признании уполномоченное лицо на основе информации о системе качества производителя должно установить, что применяются эквивалентные стандарты GMP. Эту информацию, как правило, получают путем участия в аудите систем качества производителей. И в первом, и во втором случае уполномоченное лицо может выполнить оценку соответствия на основании документации, предоставленной производителем из другой страны (см. пункт 40 данного приложения);
- при ввозе препаратов сравнения, когда невозможно получить гарантию того, что каждая серия продукции была произведена в соответствии с эквивалентными стандартами GMP, уполномоченное лицо должно указать, что каждая произведенная серия прошла все необходимые виды контроля и испытаний, необходимые для подтверждения ее качества в соответствии с информацией, предоставленной спонсором компетентному органу при запросе разрешения на проведение клинических исследований****.
- В ЕС требования к выдаче разрешения на выпуск лекарственных средств, предназначенных для исследований, уполномоченным лицом изложены в ст. 13.3 Директивы 2001/20/ЕС.
- При оценке каждой серии продукции перед выдачей разрешения на выпуск следует рассматривать:
- протоколы серии, в том числе протоколы контроля качества, протоколы внутрипроизводственного контроля и документацию, свидетельствующую о соответствии досье на лекарственное средство заказу и коду рандомизации. В эти протоколы должны быть внесены все отклонения или внесенные в плановом порядке изменения, а также любые дополнительные проверки или испытания. Протоколы должны быть полными и согласованными с персоналом, уполномоченным на это в соответствии с системой качества;
- условия производства;
- данные от аттестации (испытаниях) технических средств, процессов и методов;
- проверку окончательной упаковки;
- результаты любых анализов или испытаний, проведенных после импортирования, если необходимо;
- отчеты о стабильности;
- данные о поставщике и проверке условий хранения и транспортирования;
- отчеты об аудитах системы качества производителя;
- документы, подтверждающие право производителя на производство лекарственных средств для исследований, или соответствующие документы на экспорт, выданные компетентными органами страны-экспортера;
- при необходимости, нормативные требования в регистрационной документации (лицензии) по применению стандартов GМР и любые официальные подтверждения выполнения требований GМР;
- все другие факторы, которые уполномоченное лицо считает значимыми для качества серии. Значение вышеприведенных факторов зависит от страны, в которой производят препарат, предприятия-производителя, а также статуса препарата на рынке (является ли он зарегистрированным, зарегистрирован ли он в Российской Федерации или в других странах), а также от фазы разработки.
- Если лекарственные средства для исследований производят и упаковывают на разных участках, за которые несут ответственность разные уполномоченные лица, необходимо выполнять требования приложения 16 к настоящему стандарту.
- Если согласно действующему законодательству упаковка и маркировка осуществляется в исследовательском учреждении фармацевтом, проводившим клинические исследования, под его наблюдением, либо другим медицинским работником, то контроль этой деятельности не входит в обязанности уполномоченного лица. Однако спонсор несет ответственность за документальное оформление работы и выполнение ее в соответствии с требованиями GМР. По этому вопросу он должен получить информацию от уполномоченного лица.
Отгрузка
- Лекарственные средств для исследований должны оставаться под контролем спонсора до завершения двухэтапной процедуры выдачи разрешения на выпуск: оценки соответствия уполномоченным лицом и выдачи спонсором разрешения на выпуск для использования в клиническом исследовании после соблюдения установленных требований. Оба этапа должны быть оформлены документально (форма сертификата на серию приведена ниже). Документация должна храниться непосредственно у спонсора или у лица, которое действует от его имени. Спонсор должен убедиться в том, что условия заявки на проведение клинических исследований и принятые во внимание уполномоченным лицом соответствуют окончательному решению надзорного органа. Должны быть приняты меры, обеспечивающие выполнение этих требований. Лучшим способом для достижения этой цели может быть контроль внесения изменений в спецификацию на продукцию и соответствующее техническое соглашение между уполномоченным лицом и спонсором.
- Транспортирование лекарственных средств для исследований следует осуществлять в соответствии с инструкциями, предоставленными в распоряжение спонсором или лицом, действующим от его имени.
- До поставки лекарственных средств для исследований к месту проведения исследований должны быть заключены соглашения по декодированию уполномоченным на то персоналом.
- Следует хранить подробный перечень отгруженной продукции, составленный производителем или импортером. Особое внимание следует уделять точности указания наименования и адреса получателя.
- Передачу лекарственных средств для исследований из одного места проведения исследований на другое следует проводить только в исключительных случаях. Порядок передачи должен быть установлен инструкцией. Следует проверить историю лекарственного средства за тот период, когда он находился вне контроля производителя, например, с помощью отчетов о клинических исследованиях или документов об условиях хранения на предыдущем месте проведения исследований. Такая проверка должна учитываться при оценке пригодности к транспортированию с учетом мнения уполномоченного лица. При необходимости, препарат следует вернуть производителю или другому имеющему на то право производителю для повторной маркировки и для его оценки уполномоченным лицом. Следует хранить протоколы и обеспечивать полное отслеживание подобных передач.
Рекламации
- Результаты любого расследования, связанного с качеством препарата, должны быть рассмотрены производителем или импортером и спонсором. В этом должны участвовать уполномоченное лицо и лица, ответственные за проведение клинических исследований, чтобы оценить возможное влияние на испытания, разработку препарата и субъектов исследований.
Отзывы и возвраты Отзывы
- Порядок возврата лекарственных средств для исследований и его документального оформления должен быть согласован между спонсором и производителем или импортером. Исследователь и другие лица должны понимать свои обязанности при выполнении возврата.
- Спонсор должен убедиться в наличии у поставщика препарата сравнения или других лекарств, используемых в клинических исследованиях, а также системы для извещения спонсора о необходимости отзыва любой серии препарата.
Возвраты
- Лекарственные средства для исследований следует возвращать в соответствии с требованиями спонсора и инструкцией.
- Возвращенные лекарственные средства для исследований должны быть четко обозначены. Их следует хранить в специально отведенной контролируемой зоне. Следует сохранять документацию на возвращенные лекарственные средства.
Уничтожение
- Спонсор несет ответственность за уничтожение неиспользованных и/или возвращенных лекарственных средств для исследований. Не допускается уничтожение лекарственных средств для клинических исследований без получения разрешения от спонсора.
- Для каждого места проведения исследований и каждого периода исследований спонсор или лицо, действующее от его имени, должен составлять баланс и проверить количество препарата, которое поставлено, использовано и возвращено. Уничтожение неиспользованных лекарственных средств для исследований для данного места проведения исследований или данного периода исследований следует осуществлять только после проведения расследования, предоставления объяснений любых несоответствий и составления баланса. Документальное оформление операций по уничтожению препарата необходимо вести таким образом, чтобы можно было представить отчет о всех операциях. Протоколы следует хранить у спонсора.
- В случае уничтожения препаратов, спонсору должно быть представлен акт с указанием даты или другой документ об уничтожении. В этих документах следует четко указать номера серий и/или номера пациентов (или обеспечить возможность их отслеживания) и количество уничтоженных препаратов.
| а) Наименование (имя), адрес и телефон спонсора, исследовательской организации или исследователя, работающего по контракту (основной контакт для получения информации о препарате, клинических исследованиях и экстренном декодировании); b) лекарственная форма, способ введения, количество дозированных единиц и в случае открытого исследования наименование/шифр лекарственного средства и его активность/эффективность; с) номер серии и/или код для идентификации содержимого и операции по упаковке; d) номер (код) исследования, позволяющий идентифицировать исследование, медицинское учреждение, исследователя и спонсора, если все это не указано в другом месте; е) идентификационный номер/лечебный номер субъекта клинического исследования и при необходимости номер посещения; f) фамилия и инициалы исследователя (если не указано в пунктах а) или d) таблицы); g) инструкции по применению (может быть приведена ссылка на листок-вкладыш либо другой пояснительный документ, предназначенный для субъекта клинических исследований или лица, которое вводит препарат); h) надпись «Только для клинических испытаний» или аналогичная формулировка; i) условия хранения; j) срок использования с указанием месяца и года таким образом, чтобы избежать любой неопределенности (может быть указана дата, до которой необходимо использовать препарат, срок годности или дата повторного контроля); k) надпись «Хранить в недоступном для детей месте» за исключением случаев, когда препарат предназначен для использования только в стационаре. |
Общий случай Для первичной и наружной упаковок (пункт 26) Информация, указанная в пунктах а1k таблицы |
||
|
Первичная упаковка Если первичную упаковку и вторичную упаковку хранят вместе (пункт 29)5 a2 b3 c d e |
|||
| Первичная упаковка Блистеры или упаковки малого размера (пункт 30)5 | |||
| a2 b3,4 c d e | |||
| 1 Адрес и номер телефона основного контактного лица для передачи информации относительно препарата, клинического исследования и срочного декодирования может быть не указан на этикетке, если субъекту исследований предоставлены листок-вкладыш или карточка, на которой указаны эти данные, и дана инструкция держать их при себе постоянно (см. пункт 27 данного приложения). 2 Адрес и номер телефона основного контактного лица для передачи информации относительно препарата, клинического исследования и экстренного декодирования на этикетке не указывают. 3 Способ введения допускается не указывать только для твердых лекарственных форм для орального применения. 4 Допускается не указывать лекарственную форму и количество дозированных единиц. 5 Если на вторичной упаковке содержится информация, приведенная в пункте 26 данного приложения. | |||
Правила GMP EC 2019
Таблица 13.2 — Информация о выдаче разрешения на выпуск серии лекарственного средства для исследований| Учитываемые факторы(3) | Импортируемый препарат | ||||
| Препарат, произведенный в РФ без лицензии | Препарат, произведенный в РФ при наличии лицензии | При отсутствии лицензии | При наличии лицензии | Препарат сравнения, когда не может быть получена документация, подтверждающая, что он произведен в соответствии с установленными требованиями | |
| До проведения клинических исследований | |||||
| a) Условия транспортирования и хранения | Да | ||||
| b) Факторы(1), подтверждающие, что каждая серия произведена и разрешена для выпуска в соответствии с установленными требованиями или правилами GМР. | Да − | Да (2) | |||
| c) Документация, свидетельствующая, что каждая серия была разрешена для выпуска в соответствии с требованиями GМР. | Да | ||||
| d) Документация, подтверждающая соответствие препарата установленным требованиям к регистрации и выдаче разрешения на выпуск. | Да | ||||
| e) Результаты всех проверок и испытаний, осуществляемых для оценки качества ввезенной серии, в соответствии с требованиями, установленными при регистрации или досье на лекарственное средство. Если эти проверки и испытания проведены вне Российской Федерации, следует представить необходимые документы, а уполномоченное лицо должно указать, что они были проведены в соответствии с требованиями GМР, как минимум, эквивалентными настоящему стандарту. | − Да Да | Да − Да | − Да Да | ||
| После проведения клинических исследований | |||||
| f) В дополнение к оценке, осуществляемой перед проведением клинических исследований, все дополнительные факторы(1), подтверждающие, что каждая серия была кодирована, упакована в специальную упаковку для исследования, маркирована и испытана в соответствии с установленными требованиями или правилами GМР, как минимум эквивалентными настоящему стандарту. | Да − | Да (2) | |||
| (1) Эти факторы приведены в пункте 40 настоящего приложения. (2) Если имеется соглашение о взаимном признании или другое соглашение и применяются эквивалентные нормы GMP. (3) Должны быть учтены уполномоченным лицом, которое оценивает соответствие серии перед выдачей разрешения на выпуск. | |||||
Содержание сертификата серии
- Наименование продукта (продуктов) в соответствии обозначениями в заявке на клинические исследования, если требуется.
- Кодовое обозначение в соответствии с установленными требованиями и номер кода спонсора, если он есть.
- Эффективность
- Дозированная форма (фармацевтическая форма).
- Размер упаковки (содержимого в контейнере) и ее тип (например, флаконы, бутылки, блистеры).
- Номер серии
- Срок годности или дата повторного контроля.
- Наименование и адрес производителя, по которому расположено уполномоченное лицо, выдавшее сертификат.
- Номер регистрационного досье производителя для площадки, указанной в п.
- Примечания.
- Любая дополнительная информация по усмотрению уполномоченного лица.
- Заключение о сертификации.
- «Настоящим я удостоверяю, что серия соответствует требованиям ….».
- Фамилия, имя и отчество уполномоченного лица, выдавшего сертификат.
- Подпись
- Дата
Общие положения
Ионизирующее излучение может использоваться в производственном процессе для снижения бионагрузки, стерилизации исходных материалов, упаковки или изделий, обработки препаратов на основе крови и других целей. Существует два вида радиационных установок: с радионуклидными источниками гаммаизлучения и с ускорителями электронов высокой энергии. Источники гамма-излучений: используются два режима обработки: а) порционный (по сериям): серия продукции размещается в фиксированном положении вокруг источника излучения и не может быть загружена или выгружена во время облучения;- b) непрерывный: автоматизированная транспортная система доставляет продукцию в камеру для облучения, перемещает ее с установленной скоростью по заданному маршруту, а затем выводит из камеры.
Ответственность
- Радиационную обработку продукции может проводить непосредственно ее производитель или по контракту организация, имеющая в распоряжении радиационную установку. При этом они должны иметь разрешения, оформленные в установленном порядке.
- Производитель фармацевтической продукции несет ответственность за качество продукции, в т. ч. за результаты воздействия ионизирующего излучения. Организация, проводящая радиационную обработку, несет ответственность за то, чтобы каждая упаковка получила дозу, определенную производителем (в т. ч. упаковка с продукцией, самая удаленная от источника излучения).
- Требуемая поглощенная доза с указанием установленных предельных значений должна быть указана в документации на продукцию (регистрационном досье).
Дозиметрия
- Дозиметрия — измерение поглощенной дозы ионизирующего излучения с помощью дозиметров. Понимание принципов работы и правильное использование техники имеют важное значение для аттестации (испытаний), наладки и контроля процесса.
- Калибровка каждой партии рабочих дозиметров должна быть прослеживаемой вплоть до национального или международного эталонов. Следует строго соблюдать установленную периодичность калибровки.
- Для определения изменения поглощения штатных дозиметров после облучения как при калибровке, так и при проведении дозиметрии должен использоваться один и тот же прибор. При использовании разных приборов они должны быть калиброваны в абсолютных единицах поглощения.
- В зависимости от типа используемых дозиметров необходимо учитывать возможные источники погрешности, зависящие от влажности, температуры, периода времени между облучением и измерением, а также мощности поглощенной дозы.
- Приборы, используемые для измерения изменения поглощения дозиметров по длине волны и для измерения толщины дозиметров, необходимо калибровать. Периодичность калибровки зависит от назначения, стабильности и способа применения используемого прибора.
Аттестация (испытания) процесса
- Аттестация (испытания) – действие, доказывающее, что процесс, т.е. получение продукцией заданной поглощенной дозы, достигает ожидаемых результатов. Требования к аттестации (испытаниям) приведены в руководствах по использованию ионизирующего излучения в производстве лекарственных средств*.
- Аттестация (испытания) должна включать в себя составление карты дозного поля, чтобы установить распределение поглощенной дозы внутри облучаемого контейнера с продукцией при определенной конфигурации упаковки продукции.
- Спецификации на процесс облучения должны включать в себя:
- описание упаковки продукции;
- конфигурацию(и) укладки продукции внутри контейнера. Если в контейнере находится продукция различных видов, необходимо обратить особое внимание на возможное недооблучение продукции с высокой плотностью или затенение других изделий такой продукцией. Каждый способ укладки в контейнер разных видов продукции должен быть описан в технической документации и аттестован;
- конфигурацию расположения контейнеров вокруг источника (порционный режим) или маршрут облучаемых объектов внутри камеры для облучения (непрерывный режим);
- верхнее и нижнее предельно допустимые значения поглощенной дозы в продукции (и соответствующие показания дозиметров);
- верхнее и нижнее предельные значения поглощенной дозы в продукции по всему объему контейнера и соответствующие методы дозиметрии для контроля этих значений;
- другие параметры процесса, в т. ч. мощность поглощенной дозы, максимальное время облучения, количество циклов облучения и т. д.
Аттестация установки
Общие положения
- Аттестация заключается в экспериментальном получении и документировании фактов, доказывающих, что радиационная установка способна в течение длительного времени функционировать в установленных пределах согласно документации на процесс. Согласно данному приложению, установленные пределы — максимальное и минимальное допустимые значения поглощенной дозы в продукции (по всему объему облучаемого контейнера). Изменения в работе установки, которые могут привести к выходу значений поглощенной дозы за эти пределы, ни при каких условиях не должны происходить без ведома оператора.
- Аттестация должна включать в себя:
- основные параметры установки;
- карту дозного поля;
- документирование;
- требования к повторному вводу установки в эксплуатацию.
Источники гамма-излучения Основные параметры
- Поглощенная доза в продукции зависит от следующих основных факторов:
- активности и геометрии источника излучения;
- расстояния от источника до контейнера;
- продолжительности облучения, контролируемой таймером или скоростью движения конвейера
- состава и плотности материала, а также наличия другой продукции между источником и рассматриваемой частью контейнера.
- Суммарная поглощенная доза зависит также от маршрута, по которому движутся контейнеры при непрерывном режиме облучения, или от конфигурации загрузки при порционном режиме облучения и количества циклов облучения.
- При фиксированном маршруте (при непрерывном облучении) или при фиксированной конфигурации загрузки (при порционном режиме облучения), а также для постоянной мощности источника и вида продукции основным параметром установки, контролируемым оператором, является скорость конвейера или время, установленное на таймере.
Карта дозного поля
- При определении карты дозного поля камера для облучения должна быть заполнена контейнерами с муляжами или образцами продукции однородной плотности. Дозиметры должны быть расположены не менее чем в трех заполненных контейнерах, окруженных аналогичными контейнерами или муляжами продукции. Если продукция уложена неравномерно, дозиметры должны быть размещены в большем количестве контейнеров.
- Расположение дозиметров зависит от размеров облучаемого контейнера. Например, для контейнеров размером 1,0 х 1,0 х 0,5 м дозиметры могут располагаться дозиметров в узлах трехмерной решетки с шагом 20 см, с учетом внешней поверхности контейнера. Если предполагаемые зоны с максимальной и минимальной дозами известны из предыдущих опытов, то часть дозиметров может быть изъята из зон со средними значениями доз и помещена в зоны с экстремальными значениями дозы с шагом 10 см.
- В результате этой процедуры должны быть определены минимальная и максимальная поглощенные дозы, полученные продукцией в контейнере и на его поверхности для заданной комбинации параметров установки и облучения, плотности и конфигурации укладки.
- В идеальном случае для определения карты дозного поля следует использовать эталонные дозиметры, поскольку они имеют наибольшую точность. Допускается использовать штатные дозиметры. При этом в зонах с предполагаемыми максимальными и минимальными значениями поглощенной дозы облучения, а также в точках, предназначенных для контроля процесса, возле штатных дозиметров рекомендуется помещать эталонные дозиметры. Полученные значения поглощенной дозы будут иметь случайную погрешность, которая может быть определена путем многократных измерений.
- Минимальная наблюдаемая доза, измеряемая штатными дозиметрами, необходимая для получения всеми контейнерами минимальной требуемой дозы, должна быть установлена с учетом случайной погрешности используемых штатных дозиметров.
- Во время определения карты дозного поля параметры установки необходимо поддерживать постоянными, контролировать и регистрировать. Эти записи вместе с результатами дозиметрии и другими полученными протоколами следует сохранять.
Радиационные установки с ускорителями электронов Основные параметры установки
- Поглощенная доза ионизирующего излучения в продукции зависит от следующих основных факторов:
- характеристики пучка (энергии электронов, среднего тока пучка, ширины развертки и неравномерности пучка по ширине развертки);
- скорости конвейера;
- состава и плотности продукции;
- состава, плотности и толщины материала, находящегося между выходным окном и облучаемой частью продукции;
- расстояния от выходного окна до контейнера.
- Основными параметрами, контролируемыми оператором, являются характеристики пучка и скорость конвейера.
Карта дозного поля
- При определении карты дозного поля дозиметры следует располагать между слоями гомогенного поглотителя, имитирующего реальную продукцию, или между слоями реальной продукции однородной плотности так, чтобы не менее 10 измерений были проведены в пределах максимального пробега электронов (пункты 18–21 данного приложения).
- При определении карты дозного поля по объему облучаемого объекта параметры радиационной установки необходимо поддерживать постоянными, контролировать и регистрировать. Эти записи следует сохранять вместе с результатами дозиметрии и другими данными.
Повторный ввод установки в эксплуатацию
- Процедура повторного ввода в эксплуатацию должна проводиться заново каждый раз, когда происходят изменения процесса или параметров радиационной установки, способные привести к изменению распределения поглощенной дозы в контейнере для облучения (например, при замене облучателя радиационной установки). Объем работ по повторному вводу в эксплуатацию зависит от степени изменений, внесенных в конструкцию облучателя радиационной установки или конфигурацию загрузки. При наличии сомнений процедуру повторного ввода установки в эксплуатацию следует провести заново.
Помещения
- Во избежание перекрестного загрязнения в помещении должны быть предусмотрены меры по разделению облученных и необлученных контейнеров. Если материалы находятся в закрытых контейнерах и отсутствует риск перекрестного загрязнения, нет необходимости при облучении отделять фармацевтические материалы от нефармацевтических. Любая возможность загрязнения продукции радионуклидами должна быть исключена.
Технологический процесс
- Контейнеры с продукцией следует устанавливать в соответствии с конфигурацией загрузки, определенной в процессе аттестации (испытаний).
- В ходе облучения поглощенная доза на поверхности контейнера должна контролироваться в соответствии с аттестованными (испытанными) дозиметрическими методиками. Зависимость между этой дозой и дозой, поглощенной продукцией внутри контейнера, должна быть установлена при аттестации (испытаниях) радиационной установки.
- Для того чтобы различать облученные и необлученные контейнеры, допускается использование индикаторов ионизирующего излучения. Однако их не следует использовать в качестве единственного средства, определяющего правильность проведения процесса.
- Одновременную обработку разных видов продукции в одной загрузке следует проводить только тогда, когда по результатам эксплуатации установки или по другим данным установлено, что поглощенная доза в каждом виде продукции находится в установленных пределах.
- Получение требуемой поглощенной дозы облучения за несколько циклов излучения должно быть согласовано между производителем и организацией, проводящей облучение. Полная доза облучения должна быть получена в течение предварительно установленного интервала времени. Производитель должен быть уведомлен о фактах незапланированных перерывов между циклами облучения, если продолжительность перерывов превышает ранее согласованные значения.
- Облученная продукция всегда должна быть отделена от необлученной. Способы достижения этого включают в себя использование индикаторов радиации (пункт 31) и соответствующей планировки помещений (пункт 28).
Гамма-излучение
- При непрерывном режиме облучения дозиметры должны быть расположены таким образом, чтобы под воздействием излучения одновременно находились не менее двух дозиметров.
- При порционном режиме в точке контроля минимального значения поглощенной дозы облучения должны быть расположены не менее двух дозиметров.
- При непрерывном режиме облучения должна быть предусмотрена индикация требуемого рабочего положения источника. Положение источника и движение конвейера должны быть связаны блокировкой. Скорость движения конвейера необходимо постоянно контролировать и регистрировать. 38 При порционном режиме облучения (по сериям) перемещение источника и продолжитель-
- Следует корректировать время облучения и скорость движения конвейера с учетом распада или дозарядки источника излучения. Периодичность аттестации (испытаний) параметров установки или скорости конвейера должна соответствовать параметрам излучателя.
Радиационная установка с ускорителем электронов
- Дозиметр должен быть размещен на каждом контейнере в контрольной точке.
- Необходимо непрерывно регистрировать среднее значение тока пучка, энергию электронов, ширину развертки и скорость конвейера. Эти параметры, за исключением скорости конвейера, необходимо проверять через определенные интервалы времени, установленные при аттестации, поскольку они подвержены спонтанному изменению.
Документация
- Количество поступивших контейнеров и контейнеров, прошедших облучение и вывезенных с предприятия, должно совпадать и соответствовать значениям, указанным в сопроводительной документации. Любые расхождения должны протоколироваться и расследоваться.
- Оператор радиационной установки должен подтверждать в письменном виде диапазон значений поглощенных доз, полученных каждым контейнером при каждой загрузке или в серии продукции.
- Технологические протоколы и протоколы контроля для каждой серии продукции, прошедшей облучение, должны проверяться и подписываться специально назначенным лицом и храниться на предприятии. Метод и место хранения должно быть определено по договоренности между организацией, проводившей облучение, и производителем.
- Документация, относящаяся к аттестации (испытаниям) радиационной установки, должна храниться в течение одного года после истечения срока годности или, по крайней мере, в течение пяти лет после выпуска последней продукции, прошедшей облучение на этой установке, в зависимости от того, какой период дольше.
Микробиологический контроль
- Ответственность за проведение микробиологического контроля, в т. ч. за контроль окружающей среды в месте производства продукции и контроль продукции перед облучением, проводимый в соответствии с нормативной документацией, несет производитель лекарственных средств.
Основные принципы
Данное приложение применяется ко всем типам компьютеризированных систем, используемых в области применения настоящего стандарта. Компьютеризированная система представляет собой набор программных компьютерное оборудование, которые совместно выполняют определенные функции. Применение компьютеризированной системы и информационно-технологическая инфраструктура должны быть аттестованы. Если компьютеризированная система заменяет ручное управление, это не должно приводить к снижению качества продукции, технологического контроля или обеспечения качества. Общий риск процесса не должен возрастать.Общие положения
1 Анализ рисков
Анализ рисков должен выполняться в течение жизненного цикла компьютеризированной системы в целях обеспечения безопасности пациентов, целостности данных и качества продукции. Решения по объему испытаний и контролю целостности данных должны основываться на обоснованной и документально оформленной оценке рисков компьютеризированной системы.2 Персонал
Следует организовать взаимодействие между всеми лицами, имеющими отношение к данному процесс, включая владельцев процесса и системы, уполномоченных лиц и информационнотехнологический персонал. Весь персонал должен иметь необходимую квалификацию, уровень доступа и нести ответственность за выполнение своих обязанностей.3 Поставщики продукции и услуг
- Если для поставки, установки, наладки, задания конфигурации, интегрирования, аттестации, технического обслуживания (в том числе через удаленный доступ), модификации или поддержания компьютеризированных систем, оказания связанных с ними услуг или обработки данных привлекаются третьи лица (в частности, поставщики продукции и услуг), то между производителем и указанными третьими лицами заключаются договоры. Такие договоры должны предусматривать ответственность третьих лиц за выполнение своих обязанностей. Аналогичные требования предъявляются к информационно-технологическим подразделениям.
- Основными требованиями к поставщикам продукции или услуг являются их компетентность и надежность. Необходимость в аудите рекомендуется определять с использованием оценки рисков.
- Документация, прилагаемая к коммерчески доступным готовым для использования программным продуктам, должна быть рассмотрена пользователями, которые подлежат контролю, на предмет соответствия требованиям производителя.
- Информация о системе качества и аудитах поставщиков или разработчиков программного обеспечения и установленных систем должна быть доступна для предоставления лицам, осуществляющим контроль, по их требованию.
Стадия разработки
4 Аттестация
- Документация аттестации я и отчеты должны охватывать соответствующие стадии жизненного цикла компьютеризированной системы. Производитель должен обосновать свои стандарты, протоколы, критерии приемлемости, инструкции и записи на основе оценки рисков.
- Документация по аттестации должна включать протоколы контроля изменений (если требуется) и отчеты о любых отклонениях, выявленных при аттестации.
- Следует иметь действующий перечень (реестр) всех используемых компьютеризированных систем с указанием их функций в рамках области применения настоящего стандарта.
- Спецификации требований пользователя (задание на разработку) должны описывать необходимые функции системы на основе документально оформленной оценки рисков и соблюдения настоящего стандарта. Требования пользователя должны прослеживаться в течении всего жизненного цикла системы.
Правила GMP EC 2019
- Пользователь должен предпринять все меры, гарантирующие, что компьютеризированная система разработана в соответствии с системой обеспечения качества. Поставщик должен быть оценен соответствующим образом.
- Для аттестации компьютеризированных систем, разработанных по индивидуальному заказу или модифицированных в соответствии с требованиями заказчика, необходимо разработать методику оценки качества и эксплуатационных характеристик системы на всех этапах ее жизненного цикла с оформлением соответствующих отчетов.
- Следует предоставить свидетельства соответствия методик контроля и тестирования. В частности, следует пределы параметров системы (процесса), границы данных и действия в случае ошибок. Необходимо документально оформить оценку соответствия автоматизированных средств тестирования и режимов их работы.
- Если данные переводятся в другой формат или систему данных, то аттестация должна включать проверку неизменности значения и/или смысла данных в процессе их переноса.
Стадия эксплуатации
5 Данные
Компьютеризированные системы, осуществляющие электронный обмен данных с другими системами, должны иметь соответствующие встроенные средства контроля правильного и безопасного ввода и обработки данных с целью минимизации рисков.6 Контроль точности
Для критических данных, вводимых вручную, необходимо предусмотреть дополнительный контроль точности ввода данных. Этот контроль может осуществляться вторым оператором или с помощью аттестованных электронных средств. Критичность и возможные последствия ошибок или неправильного ввода данных в систему должны быть учтены при анализе рисков.7 Хранение данных
- Следует защищать от искажений как физическими, так и электронными мерами. Следует проверять доступность, читаемость и точность сохраненных данных. Доступ к данным должен быть обеспечен на протяжении всего периода их хранения.
- Следует выполнять регулярное резервное копирование всех необходимых данных. Сохранность и точность резервных копий, а также возможность восстановления данных должны быть проверены в процессе аттестации и периодически контролироваться.
8 Распечатки
- Следует иметь возможность получения четких печатных копий данных, хранящихся в электронном виде.
- Для записей, сопровождающих разрешение на выпуск серии, должна быть предусмотрена возможность получения распечаток, указывающих, изменялись ли какие-либо данные с момента их первоначального ввода.
9 Контрольные следы
Следует предусмотреть внесение в систему записей всех существенных изменений и удалений, связанных с применением настоящего стандарта (система, создающая «контрольные следы»), прибегая к анализу рисков. Следует таких документально оформлять причины изменений или удалений Контрольные следы должны быть доступными, должна быть возможность их преобразования в понятную для пользователей форму, их следует регулярно рассматривать.10 Внесение изменений и изменение конфигурации
Любые изменения в компьютеризированной системе, включая конфигурацию системы, должны проводиться только контролируемым способом в соответствии с инструкцией.11 Периодическая оценка
Следует периодически оценивать компьютеризированные системы ля подтверждения того, что они соответствуют результатам и настоящему стандарту. При этом следует, где требуется, оценивать текущие функциональные возможности, данные об отклонениях, сбоях, проблемах, истории обновлении, отчеты об эксплуатации, надежности, защищенности и о проведении аттестации.12 Безопасность
- Следует предусмотреть физические и логические средства защиты для ограничения доступа к компьютеризированной системе только лиц, имеющими на это право. Средства для предотвращения несанкционированного доступа к системе могут включать в себя использование ключей, карточек доступа, персональных кодов с паролями, биометрических данных, ограничения доступа к компьютерному оборудованию и зонам хранения данных.
- Степень защиты зависит от критичности компьютеризированной системы.
- Создание, изменение и аннулирование прав доступа должно быть оформлено документально.
Правила GMP EC 2019
- Следует разработать систему контроля данных и документов для идентификации операторов, которые входят в систему, и для регистрации изменения, подтверждения или удаления данных, включая дату и время.
13 Действия при сбоях в работе
Следует регистрировать и анализировать все сбои в работе, включая системные сбои и ошибки данных. Необходимо установить основную причину критических сбоев и использовать эту информацию в качестве основы корректирующих и предупреждающих действий.14 Электронная подпись
Документы в электронной форме могут быть заверены электронной подписью. Электронные подписи должны: а) в рамках организации иметь ту же силу, что и подписи от руки;- b) быть неразрывно связанными с соответствующими документами; с) включать время и дату, когда они были поставлены.
15 Выпуск серии
В случаях, когда выпуск серии продукции осуществляется с использованием компьютеризированной системы, она должна предоставлять доступ для выпуска серии только уполномоченному лицу и четко идентифицировать и регистрировать уполномоченное лицо, которое одобрило и выпустило серию. Эти действия должны осуществляться с использованием электронной подписи.16 Непрерывность работы
С целью обеспечения работоспособности компьютеризированных систем, сопровождающих критические процессы, необходимо принять меры предосторожности для гарантии непрерывности поддержки этих процессов в случае выхода системы из строя (например, с использованием ручной или дублирующей системы). Время, необходимое для введения в действие дублирующих средств, должно учитывать риски и соответствовать конкретной компьютеризированной системе и сопровождаемому процессу. Эти меры должны быть надлежащим образом оформлены документально и проверены.17 Архивирование данных
В случае необходимости архивирования данные следует проверять на доступность, удобство чтения и целостность. Если в компьютеризированной системе необходимо провести существенные изменения (например, компьютерного оборудования или программного обеспечения), следует обеспечить и проверить возможность восстановления данных.Термины и определения
Владелец процесса (process owner): лицо, ответственное за рабочий процесс; Владелец системы (system owner): лицо, ответственное за доступ и техническое обслуживание компьютеризированной системы и безопасности данных, находящихся в этой системе; Жизненный цикл (life cycle): все стадии существования компьютеризированной системы от постановки задачи до прекращения эксплуатации, включая задание исходных требований разработку, программирование, тестирование, установку, пользование и обслуживание; Информационно-технологическая инфраструктура (IT Infrastructure): компьютерное оборудование и программное обеспечение, такое как сетевые и операционные системы, которые позволяют применять их для выполнения определенных функций; Компьютеризированная система, изготовленная по индивидуальному заказу (bespoke/customized computerized system): индивидуально разработанная компьютеризированная система для обеспечения конкретного процесса; Приложение (application): программное обеспечение, установленное на определенной платформе или компьютерном оборудовании и выполняющее специальные функции; Серийное программное обеспечение (commercial off the shelf software): коммерчески доступное программное обеспечение, пригодность которого для использования продемонстрирована большим количеством пользователей.Общие положения
- Как правило, используются два метода производства и наполнения:
- двухступенчатый метод (наполнение под давлением).
- одноступенчатый метод (холодное наполнение).
Помещения и оборудование
- Производство и наполнение рекомендуется проводить, по возможности, в закрытых системах. 3 Там, где продукция или ее чистые компоненты содержатся открытыми, помещения должны соответствовать, как минимум, зоне Воздух в них должен подаваться через фильтры, а доступ осуществляться через воздушные шлюзы.
Производство и контроль качества
- Дозирующие клапаны для аэрозолей являются более сложными, по сравнению с большинством устройств, используемых в фармацевтической промышленности. Технические условия на них, методики отбора образцов и контроль дозирующих клапанов должны соответствовать требованиям, предъявляемым к качеству такой продукции. Особое значение имеет проведение аудита системы обеспечения качества у производителя дозирующих клапанов.
- Все летучие вещества (например, жидкие или газообразные пропелленты) должны быть пропущены через фильтр, удерживающий частицы с размерами более 0,2 мкм. Рекомендуется, по возможности, проводить дополнительную фильтрацию непосредственно перед наполнением.
- Упаковки и клапаны должны проходить очистку по аттестованной (испытанной) методике, учитывающей свойства продукции и обеспечивающей отсутствие загрязнения вспомогательными технологическими средствами (например, смазочными), а также микробного загрязнения. После очистки клапаны следует хранить в чистых закрытых упаковках. Во избежание внесения загрязнения при последующих операциях (например, при отборе проб для проведения испытаний) необходимо принимать меры предосторожности. Упаковки должны поступать на линию наполнения очищенными или очистка должна проводиться на линии непосредственно перед наполнением.
- Необходимо обеспечить однородность суспензии в точке наполнения в ходе всего процесса наполнения.
- При использовании двухступенчатого метода для получения заданной смеси необходимо обеспечить точную массу вводимых веществ на обоих этапах. Поэтому во многих случаях целесообразен 100 %-ный контроль массы на каждом этапе.
- Контроль после наполнения должен подтвердить отсутствие утечек. Все проверки на наличие утечек следует проводить так, чтобы не допустить микробного загрязнения или остаточной влаги.
Общие положения
Жидкости, кремы и мази могут быть особенно предрасположенными к микробному и иному загрязнению. Поэтому необходимо принимать специальные меры по предупреждению любого вида загрязнения.Помещения и оборудование
- Для защиты от загрязнения при производстве и перемещении продукции рекомендуется использовать закрытые системы. Производственные зоны, в которых находится открытая продукция или открытые чистые упаковки, как правило, следует оборудовать эффективной системой вентиляции, имеющей фильтры очистки воздуха.
- Конструкция и расположение реакторов, емкостей, трубопроводов и насосов должны предусматривать удобство их очистки и, при необходимости, дезинфекции. В частности, в конструкции оборудования должно быть сведено к минимуму наличие недоступных зон и тупиков, в которых могут концентрироваться остатки материалов, вызывающие размножение микроорганизмов.
- По возможности, не следует использовать оборудование из стекла. Как правило, детали оборудования, входящие в контакт с продукцией, должны быть изготовлены из высококачественной нержавеющей стали.
Производство
- Должны быть установлены требования к химическому и микробиологическому качеству воды, используемой в производстве, а также должен обеспечиваться контроль выполнения этих требований. Во избежание риска размножения микроорганизмов следует надлежащим образом организовать обслуживание систем подготовки воды. После обработки систем подготовки воды химическими средствами их необходимо промыть по аттестованной методике, гарантирующей полное удаление дезинфицирующих веществ.
- Качество материалов, получаемых в цистернах, следует проверять до их перемещения в емкости для хранения.
- Следует контролировать передачу материалов по трубопроводам, чтобы гарантировать их поступление в нужное место.
- В помещениях, где содержится открытая продукция или чистые упаковки, не допускается нахождение материалов, способствующих выделению волокон и других загрязняющих веществ (например, картонных или деревянных поддонов).
- Во время операций наполнения следует обеспечивать однородность (гомогенность) смесей, суспензий и т. п. Процесс перемешивания и наполнения подлежит аттестации (испытаниям). Особое внимание необходимо уделять обеспечению однородности смеси в начале, после остановок и в конце процесса наполнения.
- Если готовый продукт упаковывается не сразу после окончания производственных операций, необходимо установить максимально допустимое время до его упаковки и соответствующие условия хранения.
Общие положения
Отбор проб является важной операцией, при которой берется только малая часть материалов, предназначенных для производства всей серии продукции. Непредставительная проба не позволяет дать гарантированное заключение о качестве всей серии. Правильный отбор проб является одним из основных компонентов системы обеспечения качества. Примечание – Порядок отбора проб приведен в 6.11–6.14 Части I настоящего стандарта. В настоящем приложении приведены дополнительные указания по отбору проб исходных и упаковочных материалов.Персонал
- Персонал, занимающийся отбором проб, должен проходить начальное и систематическое повторное обучение по вопросам отбора проб. Программы обучения должны включать в себя:
- план отбора проб;
- письменные инструкции по отбору проб;
- методы и оборудование, используемые при отборе проб;
- методы предотвращения перекрестного загрязнения;
- меры предосторожности при работе с нестабильными и/или стерильными веществами;
- оценку внешнего вида материалов, упаковок и этикеток;
- порядок документального оформления любых непредвиденных или необычных обстоятельств.
Исходные материалы
- Подлинность всей серии исходных материалов может быть гарантирована, как правило, только при взятии проб из всех упаковок и проведении теста на подлинность каждой пробы. Допускается отбор проб из части упаковок при наличии аттестованной методики, исключающей возможность неправильной маркировки любой упаковки с исходными материалами.
- При аттестации такой методики следует учитывать:
- данные о производителе и поставщике, а также уровень выполнения ими требований настоящего стандарта;
- систему обеспечения качества предприятия-производителя исходных материалов;
- условия производства и контроля исходных материалов;
- характер и свойства исходных материалов, а также получаемой из них продукции.
- исходные материалы поступают от одного производителя или с одного предприятия;
- исходные материалы поступают непосредственно от производителя или в опечатанной производителем упаковке, причем этот поставщик имеет безупречную репутацию, а аудит его системы обеспечения качества регулярно проводится покупателем (производителем лекарственных средств) или официально уполномоченным лицом.
- исходные материалы поступают от посредников и производитель неизвестен или не прошел аудит;
- исходные материалы используются для производства парентеральной продукции.
- Качество серии исходных материалов может быть оценено путем отбора репрезентативной пробы и проведения испытаний. Для этой цели могут использоваться пробы, взятые для проведения теста на подлинность. Количество образцов, отбираемых для получения репрезентативной пробы, определяется статистическим методом и указывается в плане отбора проб. Количество отдельных образцов, которые могут быть смешаны для получения общей пробы, определяется с учетом вида материала, данных о поставщике, а также однородности этой общей пробы.
Упаковочные материалы
- При составлении плана отбора проб упаковочных материалов необходимо принимать во внимание, как минимум, их количество, требуемое качество, характер материалов (например, первичные упаковочные материалы и/или печатные упаковочные материалы), методы производства, а также информацию о системе обеспечения качества у производителя упаковочных материалов, полученную при проведении аудита. Количество отбираемых проб определяется статистически и указывается в плане отбора проб.
Общие положения
Лекарственные средства из растительного сырья имеют сложную природу и разнообразные характеристики, в связи с чем при их производстве особую роль играет контроль исходных материалов, условий хранения и переработки. Исходными материалами при производстве лекарственных средств из растительного сырья1 могут быть необработанные растения, растительное сырье2 или промежуточные продукты. Растительное сырье должно иметь требуемое качество и сопровождаться документацией для производителя промежуточных продуктов и лекарственных средств из растительного сырья. Для того чтобы убедиться в однородности состава растительного сырья, может потребоваться более подробная информация о способе его получения (выращивания). Важными факторами, влияющими на качество растительного сырья, являются выбор семян, условия выращивания и сбора. Эти факторы могут оказать влияние на стабильность готового продукта. Рекомендации в отношении системы обеспечения качества при выращивании и заготовке растений приведены в руководстве НМРС* «Руководство по выращиванию и сбору исходных материалов растительного происхождения (Guideline on Good Agricultural and Collection Practice for starting materials of herbal origin). Данное приложение распространяется на все исходные материалы растительного происхождения: лекарственные растения, растительное сырье и промежуточные продукты из растительного сырья. Иллюстрация применения настоящего стандарта (правил GMP) к производству лекарственных средств из растительного сырья приведена в таблице3.| Стадия производства | Правила выращивания и сбора растений (GACP)4 | Часть II стандарта | Часть I Стандарта |
| Выращивание, сбор и заготовка растений, морских водорослей, грибов и лишайников и сбор экссудатов (выделений) | |||
| Резка и сушка растений, водорослей, грибов, лишайников и экссудатов (выделений)* | |||
| Отжим из растений и перегонка** | |||
| Измельчение, переработка экссудатов, экстракция из растений, разделение на фракции, очистка, концентрация или ферментация субстанций из растительного сырья | |||
| Дальнейшая переработка в готовую продукцию, включая упаковывание готового лекарственного средства |
- Производитель должен убедиться, что эти стадии соответствуют требованиям, установленным при госу-
Правила GMP EC 2019
дарственной регистрации. Для стадий, выполняемых в полевых условиях (в соответствии с указанными выше требованиями), следует, при необходимости, руководствоваться Правилами выращивания и сбора растений (GACP). На последующих стадиях резки и сушки следует руководствоваться правилами GMP. ** Экстракцию из растений и дистилляцию, если они входят в перечень заготовительных операций для обеспечения соответствия продукции требованиям спецификаций, допускается проводить в полевых условиях. При этом растения должны быть выращены в соответствии с требованиями GACP. Это допускается в исключительных случаях и должно быть отражено в требованиях, установленных при государственной регистрации. Для операций, выполняемых в полевых условиях, должно быть обеспечено ведение документации, контроль и аттестация в соответствии с требованиями GMP. Контролирующие органы могут осуществлять надзор за этими операциями.Помещения и оборудование
Складские зоны
- Растительное сырье следует хранить в отдельных зонах. Эти зоны должны быть защищены от проникания в них насекомых и животных, особенно грызунов. Следует предусмотреть эффективные меры против распространения животных и микроорганизмов, привносимых с растительным сырьем, для предотвращения ферментации или роста плесени, а также против перекрестного загрязнения. Карантинное хранение поступившего растительного сырья и допущенного к использованию должно быть организовано в различных выгороженных зонах.
- Зоны хранения должны хорошо проветриваться. Порядок размещения упаковок не должен препятствовать свободной циркуляции воздуха.
- Особое внимание следует уделять обслуживанию и чистоте складских зон, в которых может образовываться пыль.
- Для хранения растений, экстрактов, настоек и другой продукции могут потребоваться особые условия по влажности, температуре и освещенности, которые необходимо обеспечить и контролировать.
Производственная зона
- При отборе проб, взвешивании, смешивании и других технологических операциях с растительным сырьем и промежуточным продуктом, сопровождающихся пылеобразованием, следует принимать особые меры по поддержанию чистоты, а также по предотвращению перекрестного загрязнения (удаление пыли, выделение специальных помещений и т. п.).
Оборудование
- Используемое в технологическом процессе оборудование, фильтрующие материалы и пр. должны быть совместимы с растворами экстрактов, чтобы не допустить выделение и нежелательную абсорбцию веществ, которые могут оказать влияние на продукт.
Документация
Спецификации на исходные материалы
- Производители лекарственных средств из растительного сырья должны убедиться в том, что они используют только исходные материалы растительного происхождения, соответствующие требованиям GMP и требованиям, установленным при государственной регистрации.
- Помимо данных, приведенных в настоящем стандарте (Часть I, раздел 4), в спецификации на растительное сырье и промежуточные продукты, используемые для производства лекарственных средств, следует включать:
- биноминальное наименование, принятое в ботанике с указанием, при необходимости, соответствующего классификатора (например, «Классификатор растений и животных» Карла Линнея); другую информацию, например, наименование культурного сорта растения и его хемотипическую разновидность;
- подробную информацию о происхождении растения (страна или местность, и при необходимости, культура, время и способ заготовки, использование пестицидов, возможность загрязнения радиоактивными веществами и т.д.);
- указание о том, какие части растения об использованы;
- данные о методе сушки, если используются высушенные растения;
- описание растения, а также данные его макрои микроскопического исследований;
- результаты испытаний на подлинность, в т. ч. испытания на подлинность ингредиентов с известной фармакологической активностью или маркеров. Следует провести специальные тесты, если возникает подозрение о фальсификации или подмене лекарственного сырья; для определения подлинности необходимо иметь соответствующие образцы сравнения;
Правила GMP EC 2019
- влажность растительного сырья, определенную в соответствии с нормативным документом*;
- по возможности, описание основных ингредиентов, обладающих установленной фармакологической активностью, или маркеров;
- методы определения содержания пестицидов и их допустимые концентрации в соответствии с нормативными документами или, при их отсутствии, аттестованным методом, если не предусмотрено иное*;
- испытания на загрязнение грибами и/или бактериями (в т. ч. афлатоксинами, другими микотоксинами и пест-инфестацинами) и предельные значения допустимого загрязнения;
- испытания на содержание токсичных металлов и других возможных посторонних примесей;
- испытания на включение инородных материалов;
- другие виды контроля в соответствии с нормативными документами**.
Технологические инструкции
- Технологические инструкции должны содержать описание различных операций, проводимых с растительным сырьем (очистка, сушка, измельчение, просеивание и пр.), а также данные о времени и температуре сушки и методах, используемых для контроля размеров фрагментов или частиц.
- Особое внимание следует обратить на наличие инструкций и протоколов, удостоверяющих, что каждая упаковка тщательно осмотрена на предмет определения фальсификации, подмены или присутствия посторонних веществ (частиц металла или стекла, остатков или экскрементов животных, камней, песка и т.д.) или следов гниения или разложения.
- Технологические инструкции должны также содержать порядок или способ удаления посторонних веществ и методы очистки (отбора) растительных материалов до помещения растительного сырья на хранение или до начала производства.
- Инструкции по приготовлению должны содержать подробное описание растворителей, времени и температуры экстракции, стадий концентрации и методов работы.
Контроль качества
Отбор проб
- Так как необработанное лекарственное сырье получают из отдельных растений, оно является неоднородным. Отбор проб должен проводить специально обученный персонал с соблюдением особой предосторожности. Подлинность каждой серии продукции должна быть подтверждена отдельным документом.
- Следует сохранять контрольные образцы растительного материала, особенно, если растительное сырье не описано в нормативных документах***. При изготовлении порошков следует сохранять образцы неразмолотого растительного материала.
- Персонал, выполняющий контроль качества, должен иметь специальную подготовку и опыт работы с растительным сырьем, промежуточными продуктами или лекарственными средствами из растительного сырья для проведения испытаний поставляемого растительного сырья на подлинность и наличие примесей, выявления роста колоний грибов, заражения паразитами, неоднородности и т. п. в полученном сырье.
- Подлинность и качество растительного сырья, промежуточных продуктов и лекарственных средств из растительного сырья должны контролироваться в соответствии с нормативной документацией****
- В правилах GMP EC указано «в соответствии с Европейской Фармакопеей».
Общие положения
Настоящее приложение распространяется на производство газов как фармацевтических активных субстанций (далее – активные субстанции) и производство газов для медицинского применения (далее – медицинские газы). Производство медицинских газов, относящихся к лекарственным средствам, должно соответствовать действующим нормативным правовым актам. Разграничение между производством активных субстанций производством лекарственных средств должно быть четко проведено в каждом регистрационном досье. Обычно стадии получения и очистки газов относятся к производству активных субстанций. Газы относятся к лекарственным средствам с момента первого сохранения газа, предназначенного для такого использования. Производство газов как активной субстанций должно соответствовать части II настоящего стандарта, данным приложением и другими приложениями, при необходимости. Производство медицинских газов должно соответствовать части I настоящего стандарта, данным приложением и другими приложениями, при необходимости. Непрерывные процессы допускаются в исключительных случаях, когда промежуточное хранение газа между производством активной субстанции и производством лекарственного средства невозможно. Такие процессы следует рассматривать как производство лекарственных средств, что должно быть ясно указано в регистрационном досье. Настоящее приложение не распространяется на получение и обращение медицинских газов в лечебно-профилактических учреждениях за исключением случаев, когда он рассматривается как промышленное производство. Не смотря на это, в этих целях могут использоваться необходимые части данного приложенияПроизводство газов как активных субстанций
Газы как активные субстанции могут быть получены методом химического синтеза или из естественных источников, в том числе путем очистки (например, на заводах по выделению газов из воздуха).- Технологические процессы получения газов указанными выше методами должны соответствовать части II настоящего стандарта, при этом:
- b) требования к последующему анализу стабильности газов как активных субстанций (часть II, п. 11.7 настоящего стандарта), который служит для подтверждения условий хранения сроков годности или повторного контроля (часть II, п. 11.6) не применяются в случае, если вместо первичного изучения стабильности были использованы данные, содержащиеся в научной литературе;
- Следует обеспечить непрерывный контроль газов как активных субстанций в течение непрерывного процесса их получения (например, разделением воздуха). Результаты контроля должны быть представлены в виде, позволяющем проводить анализ тенденций.
- В дополнение к изложенному:
- b) Операции наполнения баллонов или передвижных криогенных сосудов газами как активными субстанциями следует проводить в соответствии с требованиями для медицинских газов (п. п 22-27 данного приложения и требованиями главы 9 часть II настоящего стандарта).
Производство медицинских газов
Производство медицинских газов, как правило, осуществляется в закрытом оборудовании. Благодаря этому риск загрязнения газов из окружающей среды минимален. Однако существует риск перекрестного загрязнения другими газами, особенно при повторном использовании контейнеров.- Требования к баллонам также распространяются на связки (комплекты) баллонов (за исключением случаев хранения и транспортировки в специальных контейнерах).
Персонал
- Весь персонал, занятый в производстве и реализации медицинских газов, должен пройти обучение и понимать требования настоящего стандарта (правилам GMP) в отношении медицинских газов, знать критически важные аспекты и возможную опасность для пациентов, которую может представить эта продукция. Программы обучения должны распространяться и на водителей грузовиков.
- Персонал, организация, работающих по контрактам, который может оказать влияние на качество медицинских газов (например, занятый техническим обслуживанием баллонов и вентилей) должен пройти необходимое обучение.
Помещения и оборудование Помещения
- Операции наполнения, подготовки и наполнения баллонов и переносных криогенных сосудов медицинскими газами следует выполнять в зонах, отделенных от зон наполнения немедицинскими газами. Обмен баллонами (контейнерами) между этими зонами не допускается. Допускается наполнение, подготовка и наполнение другими газами в одной и той же зоне при условии выполнения требований к медицинским газам и выполнения технологических операций в соответствии с настоящим стандартом (правилами GMP).
- отдельные маркированные зоны для различных газов;
- однозначное обозначение и разделение баллонов (переносных криогенных сосудов), находящихся на разных стадиях производства (например, «Ожидает проверки», «Ожидает наполнения»,
- Пустые баллоны (переносные криогенные сосуды) после сортировки или технического обслуживания и наполненные баллоны (переносные криогенные сосуды) следует хранить под навесами, защищающими их от неблагоприятных погодных условий. Наполненные баллоны (переносные криогенные сосуды) следует хранить в условиях, гарантирующих их доставку в чистом виде и отвечающем условиям их использования.
- Производитель должен обеспечивать особые условия хранения, соответствующие требованиям регистрационного досье (например, для смесей газов, в которых происходит разделение фаз при замораживании).
Оборудование
- Газом следует наполнять только предназначенный для него баллон (контейнер), что должно обеспечиваться конструкцией оборудования. Трубопроводы, по которым проходят различные газы, не должны, как правило, иметь соединений. Если такие соединения необходимы (например, при наполнении смесями газов), следует провести аттестацию оборудования и показать, что риск перекрестного загрязнения разными газами отсутствует. В дополнение к этому распределительные коллекторы должны быть оборудованы специальными соединительными элементами. Эти соединений должны соответствовать действующим нормам. При использовании на одном участке наполнения соединений, выполненных по разным стандартам, их следует тщательно контролировать, так же как и адаптеры, применение которых необходимо в некоторых случаях для устройства обходных соединений в специфических системах наполнения.
- Резервуары для хранения и передвижные цистерны для доставки должны быть предназначены для одного вида газа определенного качества. Допускается хранить или транспортировать сжиженные медицинские газы в тех же резервуарах (контейнерах), что и такие же немедицинские газы, при условии, что качество последних, по меньшей мере, эквивалентно качеству медицинских газов и выполняются требования настоящего стандарта. В этих случаях следует провести и документально оформить анализ рисков.
- Допускается применение общей системы распределения газов с коллекторами медицинского и немедицинского назначения только при наличии аттестованного метода, при котором обратный потока газа из немедицинской системы в медицинскую исключен.
- Коллекторы для наполнения должны быть предназначены только для одного медицинского газа или для определенной смеси медицинских газов. В исключительных случаях допускается наполнение газов для других медицинских целей с использованием коллекторов, предназначенных для медицинских газов, если доказана такая возможность и такой процесс контролируется. В этих случаях качество немедицинского газа должно быть, по крайней мере, эквивалентным качеству медицинского газа и должны соблюдаться требования настоящего стандарта. Наполнение должно производиться по принципу организации производства циклами.
- Работы по ремонту и техническому обслуживанию оборудования (включая очистку и продувку) не должны влиять на качество медицинских газов.. В частности, производитель должен разработать и документально оформить мероприятия, проводимые после ремонта и технического обслуживания, сопровождающихся разгерметизацией систем. Производитель должен гарантировать, что оборудование не содержит никаких загрязнений, которые могут оказать влияние на качество готового продукта до его выпуска. Следует вести и хранить документацию о проведенных работах.
- Производитель должен разработать и документально оформить инструкцию, регламентирующую действия после возврата цистерны на участок или в организацию обеспечения медицинскими газами (после транспортирования немедицинского газа в условиях, указанных в п. 12 данного приложения, или после операций по техническому обслуживанию). Такие действия подлежат аналитическому контролю.
Документация
- Данные, включаемые в протокол на каждую серию наполненных баллонов (переносных криогенных сосудов), должны позволять проследить информацию обо всех основных параметрах соответствующих стадий наполнения для каждого наполненного баллона. Эти данные должны включать следующее (где требуется):
- b) номер серии;
- d) сведения о персонале, выполнявшем основные стадии процесса (например, очистку линий, приемку материалов, подготовку до наполнения, наполнение);
- используемое оборудование (например, коллектор для наполнения);
- количество баллонов и (или) переносных криогенных сосудов до операции наполнения, включая обозначение каждой емкости и ее геометрическую емкость;
- операции, выполненные до наполнения (п. 30 данного приложения);
- основные параметры, необходимые для обеспечения правильного выполнения операции по наполнению при стандартных условиях;
- результаты проверок фактического наполнения баллонов и/или передвижных криогенных сосудов;
- образец этикетки серии;
- спецификацию на готовый продукт и результаты контроля качества (включая данные о калибровке испытательного оборудования);
- количество отклоненных баллонов и (или) переносных криогенных сосудов с указанием их обозначений и причины отклонения;
- подробные сведения обо всех проблемах и необычных событиях, утвержденное разрешение на любое отклонение от инструкций по наполнению;
- Следует вести и хранить протоколы для каждой серии газа, предназначенного для наполнения резервуаров лечебно-профилактических учреждений. Эти протоколы должны включать следующее (где требуется):
- b) номер серии;
- d) дату и время операции по наполнению;
- данные о резервуаре (цистерне), из которой производилось наполнение, и данные о происхождении газе;
- все существенные детали при наполнении;
- спецификацию на готовый продукт и результаты контроля качества (включая данные о калибровке испытательного оборудования);
- подробные сведения обо всех проблемах и отклонениях, утвержденное разрешение на любое отклонение от инструкций по наполнению;
- разрешение уполномоченного лица на выпуск серии с датой и подписью.
Производство
Перемещение и подача криогенных и сжиженных газов- Перемещение криогенных или сжиженных газов с места первичного хранения, включая контроль перед перемещением, должно осуществляться в соответствии с аттестованными методами, разработанными с целью предотвращения возможного загрязнения. Трубопровод, по которому перемещается газ, должен быть оборудован обратным клапаном или другим аналогичным устройством. Гибкие соединения, нестационарные соединительные шланги и средства для соединения перед использованием должны быть промыты потоком соответствующего газа.
- Шланги, используемые для наполнения резервуаров и цистерн, должны быть оборудованы средствами для соединения, специально предназначенными для данного газа. Следует контролировать адаптеры для подключения резервуаров и цистерны.
- Подача газа в резервуары, содержащие аналогичный газ такого же уровня качества, может быть осуществлена при наличии положительных результатов контроля пробы подаваемого газа. Проба может быть взята как из подаваемого газа, так и из резервуара после завершения подачи газа.
Наполнение и маркировка баллонов и переносных криогенных сосудов
- До наполнения баллонов или переносных криогенных сосудов следует определить серию продукции, проверить на соответствие спецификации и получить допуск к наполнению.
- Для непрерывных процессов, указанных в разделе «Основные принципы» данного приложения, следует определить точки внутрипроизводственного контроля для проверки соответствия газа спецификациям.
- Баллоны, переносных криогенные сосуды и клапаны должны отвечать установленным техническим спецификациям и требованиям регистрационного досье. Они должны быть предназначены только для одного медицинского газа или определенной смеси медицинских газов. Баллоны должны иметь цветовую маркировку в соответствии с установленными требованиями. Для защиты от загрязнений баллоны следует оснащать клапанами удержания минимального давления с механизмами предотвращения обратного потока.
- Баллоны, переносные криогенные сосуды и клапаны должны быть проверены перед первым использованием в производстве и обслуживаться надлежащим образом. При использовании изделий медицинского назначения их техническое обслуживание следует проводить по инструкциям производителя.
- Операции по проверке и техническому обслуживанию не должны оказывать отрицательного воздействия на качество и безопасность лекарственного средства. Вода, используемая для испытаний баллонов гидростатическим давлением, должна отвечать требованиям к питьевой воде.
- Следует проверять внутреннее состояние баллонов до установки клапана, рассматривая это как часть работ по контролю и техническому обслуживанию. При этом следует убедиться в отсутствии воды или других загрязнений. Эта проверка выполняется:
- для новых баллонов, впервые использующимся для медицинских газов;
- после проведения испытаний гидростатическим давлением или эквивалентным методом с демонтажем клапана;
- каждый раз при замене клапана.
- Производитель лекарственного средства должен выполнять техническое обслуживание и ремонт баллонов, переносных криогенных сосудов и клапанов. При выполнении этих работ по договору (контракту) работы должны выполняться только исполнителями, указанными в договоре в соответствии с техническими требованиями. Следует проводить аудит исполнителей работ на предмет выполнения ими требований стандартов.
- Следует иметь систему прослеживаемости баллонов, передвижных криогенных сосудов и клапанов.
- Перед операцией наполнения следует выполнять:
- если баллон оснащен клапаном удержания минимального давления, то при отсутствии сигнала, о наличии остаточного избыточного давления, клапан следует проверить; если клапан функционирует неправильно, баллон следует отправить на техническое обслуживание;
- если баллон не оснащен клапаном удержания минимального давления и в баллоне не обнаружено остаточного избыточного давления, то его следует отправить на дополнительные испытания с целью проверки отсутствия воды или других загрязнений; дополнительные меры могут включать визуальный осмотр внутреннего состояния баллона, который проводится после очистки по аттестованному методу;
- b) проверку отсутствия любых прежних этикеток на серию;
- d) внешний визуальный осмотр каждого баллона, передвижного криогенного сосуда и клапана на наличие раковин, сварочных прожигов, других повреждений и загрязнения маслами; следует выполнить очистку, при необходимости;
- проверку даты следующего испытания клапана (для клапанов, подлежащих периодической проверке);
- проверку баллонов или переносных криогенных сосудов на предмет того, что все необходимые испытания (например, гидростатическим давлением или эквивалентным методом), которые требуется в соответствии с действующими нормами, проведены и действительны;
- проверку цветового кодирования каждого баллона в соответствии с регистрационным досье и и установленными требованиями.
- Для операции наполнения должен быть определена серия.
- Баллоны, возвращаемые на повторную заправку, должны быть тщательно подготовлены с целью минимизации риска загрязнения в соответствии утвержденными методами. Эти методы должны включать операции по откачке газа и/или продувки должны быть аттестованы.
- Следует тщательно готовить переносных криогенные сосуды, возвращаемые на повторную заправку, согласно утвержденным инструкциям, с целью сведения к минимуму риска загрязнений. В частности, переносные емкости, в которых отсутствует остаточное давление, должны быть подготовлены с использованием аттестованного метода.
- Производитель должен выполнять необходимые проверки для обеспечения правильного наполнения каждого баллона и (или) переносных криогенного сосуда.
- Следует выполнить контроль каждого наполненного баллона до установки устройства контроля первого вскрытия (п. 36 данного приложения) с использованием метода на отсутствие утечки. Метод контроля не должен приводить к загрязнению поверхности патрубка клапана баллона. Такой контроль следует проводить после отбора всех образцов для контроля качества, если требуется.
- После наполнения патрубки клапанов баллонов должны быть закрыты колпачками для защиты от загрязнения. На баллоны и переносных криогенные сосуды следует устанавливать устройства контроля первого вскрытия.
- На каждый баллон или переносных криогенный сосуд следует нанести этикетку. На этикетке следует указать номер серии и срок годности.
- При производстве медицинских газов путем смешивания двух или более различных газов (в линии для наполнения либо непосредственно в баллонах) производитель должен использовать аттестованный метод смешивания, который гарантирует, что газы должным образом смешаны в каждом баллоне, и смесь однородна (гомогенна).
Контроль качества
- Следует выполнить контроль каждой серии медицинского газа (баллоны, передвижные криогенные сосуды, резервуары в медицинских организациях) в соответствии с установленными требованиями и получить разрешение на выпуск.
- План отбора проб и объем проводимых испытаний должны отвечать следующим требованиям в отношении баллонов (если в регистрационном свидетельстве не указано иное):
- b) если только один медицинский газ наполняется в баллоны в рамках одного производственного цикла в один промежуток времени, то по крайней мере один баллон при каждом непрерывном цикле наполнения должен быть проверен на подлинность и количественное содержание. Примером непрерывного цикла наполнения является производство в течение одной смены одним и тем же персоналом с использованием одного оборудования и одной серии газа, который расфасовывался;
- d) в отношении газов, смешивание которых происходит до наполнения, следует выполнять те же принципы, что и в отношении наполнения одним газом, если на линии осуществляется непрерывный контроль «на линии» смеси газов, предназначенных для наполнения.
- В завершающие испытания переносных криогенных сосудов следует включать испытания на подлинность и количественное содержание в каждом сосуде, если иное не предусмотрено регистрационным досье. Выборочный контроль серий допускается проводить только в случае, если было доказано, что перед повторным наполнением критические характеристики остаточного газа в каждом сосуде остались неизменными.
- Не требуется проводить отбор проб после повторного наполнения криогенных сосудов пользователя в месте использования (сосуды в лечебно-профилактических учреждениях или переносных криогенные сосуды) из специально предназначенных цистерн при наличии документа, подтверждающего качество содержимого цистерны. При этом после последовательных повторных наполнений производитель должен показать, что качество газа в сосудах поддерживается на установленном уровне.
- Не требуется сохранять контрольные и архивные образцы серий продукции, если это не предусмотрено документацией.
- Не требуется проведение последующего исследования стабильности, если первичное изучение стабильности было заменено использованием данных, содержащихся в научной литературе.
Транспортирование газов
- Следует защищать наполненные газовые баллоны и переносных криогенные сосуды во время транспортирования, в частности, их следует доставлять заказчикам в чистом состоянии, соответствующем условиям их дальнейшего использования.
Термины и определения
баллон (cylinder): Транспортируемый контейнер вместимостью не более 150 л воды, находящийся под давлением (для целей данного приложения). Примечание – В настоящем приложении определение баллон подразумевает, в соответствующем контексте, понятие «связка баллонов» (упаковка). газ (gas): Вещество или смесь веществ, которые при давлении 1,013 бар (101,325 кПа) и температуре 15 °С находятся полностью в газообразном состоянии или при 50 °С давление паров превышает 3 бар (300 кПа) (ISO 10286:1996 Gas cylinders – Terminology. Bilingual edition). зона (area): Часть помещения, специально выделенная для производства медицинских газов. испытание гидростатическим давлением (hydrostatic pressure test): Испытание, проводимое с целью обеспечения безопасности в соответствии с требованиями национальных и международных норм для проверки того, что баллоны или резервуары могут выдержать высокие давления. клапан удержания остаточного давления (minimum pressure retention valve): Клапан, снабженный обратной системой, поддерживающей определенное давление (около 3—5 бар выше атмосферного давления) и предотвращающей загрязнение клапана при использовании. контейнер (container): Криогенный сосуд, цистерна, бак, баллон, связка баллонов или любая другая упаковка, которая вступает в непосредственный контакт с медицинским газом. криогенный газ (cryogenic gas): Газ, который при давлении 1,013 бар сжижается при температуре ниже минус 150 °С. криогенный сосуд (cryogenic vessel): Стационарный или переносной термически изолированный контейнер, сконструированный для хранения сжиженных или криогенных газов. Из криогенного сосуда газ отбирается в газообразном или жидком состоянии (для целей данного приложения). максимальный теоретический остаточный уровень примеси (maximum theoretical residual impurity): Примесь газа от возможного предыдущего загрязнения, оставшаяся в баллонах перед наполнением после предварительной обработки. Примечание – Вычисление максимального теоретического уровня примеси имеет значение только для сжатых газов в предположении, что эти газы ведут себя как идеальные. медицинский газ (medicinal gas): Любой газ или смесь газов, предназначенные для введения больным в терапевтических, диагностических или профилактических целях для оказания фармакологического воздействия и классифицируемые как лекарственные средства. нерасфасованный газ (bulk gas): Любой газ, предназначаемый для медицинского использования, который прошел все технологические стадии, за исключением стадии окончательной упаковки. обратный клапан (non-return valve): Клапан, который позволяет потоку проходить только в одном направлении. откачивать (evacuate): Удалять остаточный газ из контейнера с помощью вакуума. продувка (purge): Операция по опорожнению и прочистке баллона:- методом сброса и откачивания или
- методом сброса, частичного создания избыточного давления рассматриваемым газом и его последующим сбросом.
Общие положения
Производство иммунных лекарственных средств для животных имеет ряд особенностей, которые следует принимать во внимание при организации системы обеспечения качества. Из-за большого количества видов животных и сопутствующих патогенных микроорганизмов спектр продукции, производимой на предприятии, может быть очень широким при небольшом объеме производства. В связи с этим производство обычно организуется по принципу отдельных циклов. Более того, ввиду особенностей этого производства (стадии культивирования, отсутствие финишной стерилизации и т. п.) необходима тщательная защита продукции от прямого и перекрестного загрязнений. Большое внимание следует уделять защите окружающей среды, особенно при использовании патогенных или экзотических микроорганизмов. При использовании микроорганизмов, патогенных для человека, особенно тщательно следует защищать работающий персонал. С учетом этих факторов, а также нестабильности свойств, характерной для иммунных препаратов, и относительно низкой эффективности проводимых испытаний (в частности, при контроле качества готовой продукции), система обеспечения качества приобретает особенно важное значение. При производстве иммунных лекарственных средств для животных особую роль играет постоянный контроль за соблюдением всех требований, изложенных в части I настоящего стандарта и данном приложении. Следует проводить непрерывную оценку данных, получаемых в ходе контроля различных аспектов производства (оборудование, помещения, продукция и т. п.) на соответствие требованиям настоящего стандарта. Решения и действия, предпринятые на основании этой оценки, должны быть оформлены документально.Персонал
- Все сотрудники (включая персонал, занимающийся очисткой и обслуживанием), работающие в зонах производства иммунной продукции, должны пройти обучение по личной гигиене и микробиологии и дополнительное обучение в соответствии со спецификой производимой продукции.
- Лица, ответственные за производство и контроль качества, должны иметь базовую подготовку по следующим предметам (всем или некоторым): бактериологии, биологии, биометрии, химии, иммунологии, медицине паразитологии, фармации, фармакологии, вирусологии и ветеринарии, а также необходимые знания по защите окружающей среды.
- Персонал должен быть защищен от возможного заражения микроорганизмами, используемыми в производстве. При использовании микроорганизмов, известных как возбудители болезней человека, необходимо принимать особые меры по защите персонала, работающего с этими микроорганизмами или экспериментальными животными.
- Необходимо принимать соответствующие меры против переноса микроорганизмов людьми за пределы производственной зоны. В зависимости от вида микроорганизмов к таким мерам относятся полное переодевание и обязательное принятие душа перед выходом из производственной зоны.
- При производстве иммунной продукции особую важность представляет риск прямого и перекрестного загрязнений, вызываемых персоналом.
Помещения
- При проектировании помещений должна быть предусмотрена защита как продукции, так и окружающей среды. Это может быть достигнуто за счет использования изолированных, чистых, чистых/изолированных или контролируемых зон.
- Операции с живыми микроорганизмами должны проводиться в изолированных зонах. Уровень изоляции должен зависеть от патогенности микроорганизмов и от того, были ли они классифицированы как экзотические.
- Операции с инактивированными микроорганизмами следует проводить в чистых зонах. Чистые зоны следует также использовать при работе с неинфицированными клетками, выделенными из многоклеточных организмов, и в некоторых случаях при работе со средами, прошедшими стерилизующую фильтрацию.
- Операции с открытыми продуктами или компонентами первичной упаковки, которые не подлежат дальнейшей стерилизации, следует проводить в боксе (установке) с однонаправленным (ламинарным) потоком воздуха (зона А), находящемся в зоне В.
- Если производство иммунных лекарственных средств для животных находится в том же здании, то другие операции с живыми микроорганизмами (контроль качества, исследования и диагностика и т.п.) должны выполняться в отдельных изолированных помещениях. Степень изоляции зависит от патогенности данного вида микроорганизмов и от того, был ли он классифицирован как экзотический. При выполнении диагностических операций существует риск загрязнения высокопатогенными микроорганизмами. Уровень изоляции должен соответствовать всем вышеперечисленным рискам. Изоляция может потребоваться также в случаях, когда контроль качества и другие операции проводятся в зданиях, расположенных вблизи производственных зданий.
- Изолированные помещения должны быть легко дезинфицируемыми. При проектировании этих помещений следует предусматривать:
- отсутствие прямого выхода вентилируемого воздуха наружу;
- наличие вентиляции с обеспечением отрицательного перепада давления (разрежения). Воздух должен удаляться через НЕРА-фильтры. Не допускается рециркуляция воздуха в ту же зону, за исключением случаев его дополнительной очистки НЕРА-фильтрами (обычно это условие выполняется при прохождении рециркулирующего воздуха через приточные НЕРА-фильтры, которые используются для этой зоны). Поступление воздуха из одной зоны в другую допускается в том случае, если вытяжной воздух проходит через два последовательно установленных НЕРА-фильтра. При этом должны быть предусмотрены непрерывный контроль целостности первого фильтра и средства безопасного удаления вытяжного воздуха в случае повреждения этого фильтра;
- воздух, выходящий из производственных зон, в которых проводится работа с экзотическими микроорганизмами, должен проходить через два последовательно установленных НЕРА-фильтра. При этом рециркуляция воздуха между производственными зонами не допускается;
- наличие системы сбора и дезинфекции жидких отходов, включая контаминированный конденсат из стерилизаторов, биореакторов и т. п. Твердые отходы, в т. ч. трупы животных, следует дезинфицировать, стерилизовать или сжигать. Контаминированные фильтры необходимо удалять безопасным способом;
- комнаты для переодевания следует проектировать и использовать как воздушные шлюзы и оборудовать, при необходимости, умывальниками и душевыми кабинами. Схема перепада давления воздуха должна предотвращать движение воздуха между рабочей зоной и окружающей средой, а также риск загрязнения одежды, используемой вне рабочей зоны;
- для перемещения оборудования должна быть предусмотрена система воздушных шлюзов, конструкция которой должна предотвращать движение загрязненного воздуха рабочей зоны во внешнюю среду и риск загрязнения оборудования внутри шлюза. Размер шлюза должен позволять проводить эффективную деконтаминацию поверхности перемещаемых материалов. Рекомендуется устанавливать на двери шлюза таймер, позволяющий контролировать время, достаточное для проведения эффективной деконтаминации;
- во многих случаях для безопасного удаления отходов и подачи стерильных предметов необходимо использовать проходной автоклав с двумя дверями.
- Воздушные шлюзы для передачи материалов и комнаты для переодевания следует оборудовать блокирующими устройствами, препятствующими одновременному открыванию более одной двери. В комнаты для переодевания следует подавать очищенный воздух того же качества, что и для производственных зон. Они должны быть оборудованы системами удаления воздуха, обеспечивающими воздухообмен, независимый от производственных зон. Воздушные шлюзы для передачи материалов должны, как правило, вентилироваться таким же образом, однако допускается применять невентилируемые шлюзы или шлюзы, оборудованные только приточными системами.
- Стадии технологического процесса, при которых может произойти загрязнение продукции (хранение клеток, подготовка сред, культивирование вирусов и т. п.), должны выполняться в отдельных зонах. Особая предосторожность требуется при работе с животными и продуктами животногопроисхождения.
- Операции с микроорганизмами, проявляющими высокую устойчивость к дезинфекции (например, спорообразующие бактерии), до их инактивации следует выполнять в изолированных зонах, специально предназначенных для проведения таких операций.
- В производственной зоне следует одновременно проводить работы только с микроорганизмами одного вида, за исключением процессов смешивания и последующего наполнения.
- Следует предусматривать возможность проведения дезинфекции производственных зон в промежутках между производственными циклами с использованием аттестованных методов.
- Допускается производство микроорганизмов в контролируемых зонах при условии, что оно осуществляется в полностью закрытом и стерилизуемом тепловым методом оборудовании, а все соединения после завершения работы и перед разборкой также подвергаются тепловой стерилизации. Допускается сборка соединений под локальным однонаправленным (ламинарным) потоком воздуха при условии, что их количество ограничено, используются соответствующие асептические методы и отсутствует опасность утечки. Параметры процесса стерилизации, используемого перед разборкой соединений, должны пройти аттестацию (испытания) для всех используемых микроорганизмов. В пределах одной зоны разные продукты могут быть загружены в разные биореакторы только при отсутствии риска случайного перекрестного загрязнения. Работа с микроорганизмами, к которым предъявляются особые требования изоляции, должна выполняться только в специально выделенных зонах.
- В помещениях для содержания животных, предназначенных для использования (или используемых) в производстве, следует поддерживать режим изолированной и/или чистой зоны и отделять их от помещений для содержания других животных. Помещения, где содержатся животные, используемые для контроля качества продукции (в т. ч. и с применением патогенных микроорганизмов), должны быть изолированы.
- Доступ в производственные зоны может иметь только персонал, имеющий на это разрешение. Режим доступа в производственные зоны должен быть установлен в специальной инструкции.
- Техническая документация предприятия должна содержать необходимые данные о производственных помещениях.
Оборудование
- Конструкция и монтаж оборудования должны соответствовать специфическим требованиям для каждого вида продукции.
- Оборудование должно обеспечивать удовлетворительную первичную изоляцию биологических агентов. Конструкция и монтаж оборудования должны, при необходимости, обеспечивать его легкую и эффективную деконтаминацию и/или стерилизацию.
- Конструкция и монтаж закрытого оборудования, используемого для первичной изоляции биологических агентов, должны предусматривать предотвращение утечек или формирование капель и аэрозолей.
- Перед использованием оборудование следует стерилизовать предпочтительно сухим паром под давлением. Другие методы применимы в том случае, если оборудование не допускает стерилизацию паром. Важно не пропустить при обработке такие виды оборудования, как центрифуги и водяные бани.
- Конструкция оборудования должна исключать возможность перепутывания различных организмов или продуктов. Трубопроводы, клапаны и фильтры должны маркироваться в соответствии с их назначением.
- Некоторые виды оборудования (например, оборудование, требующее контроля температуры) должны быть оснащены регистрирующими устройствами и/или системами сигнализации. Во избежание отказов, совместно с анализом регистрируемых данных, следует организовать систему профилактического обслуживания.
- Загрузка лиофильных сушильных установок должна происходить в чистой/изолированной зоне. Операции по разгрузке лиофильной сушильной установки приводят к загрязнению окружающей среды. В связи с этим при использовании лиофильных сушильных установок, открывающихся с одной стороны, чистое помещение должно пройти деконтаминацию до перемещения в эту зону другой серии продукции, за исключением серий с одинаковыми организмами. Двусторонние лиофильные сушильные установки должны стерилизоваться после каждого цикла производства, если только они не открываются в чистую зону.
Животные и помещения для их содержания (виварии)
- Общие требования к содержанию животных, помещениям для них, уходу и карантину приведены в соответствующей нормативной документации.
- Виварии должны быть изолированы от других производственных помещений. Их проекты должны быть выполнены с соблюдением необходимых требований.
- Санитарное состояние животных, используемых в производстве, необходимо оценивать, контролировать и регистрировать. Правила обращения с некоторыми видами животных приведены в специальных руководствах.
- Животные, биологические агенты и проводимые испытания должны быть четко маркированы (документированы) в соответствии с установленной системой во избежание риска перепутывания и с целью контроля всех возможных видов опасностей.
Дезинфекция. Уничтожение отходов
- Дезинфекция и/или уничтожение твердых и жидких отходов могут иметь особенно важное значение при производстве иммунной продукции. В связи с этим необходим тщательный подход к методам и оборудованию, используемым для предотвращения загрязнения окружающей среды, а также к их аттестации (испытаниям).
Производство
- Из-за широкого разнообразия продукции, большого количества стадий при производстве иммунных лекарственных средств для животных и характера биологических процессов особое внимание следует уделять строгому соблюдению аттестованных методов, постоянному контролю всех технологических стадий производства и проведению внутрипроизводственного контроля. Особое внимание следует обратить на исходные материалы, среды культивирования и посевной материал.
Исходные материалы
- Требования к исходным материалам должны быть четко определены в документально оформленных спецификациях. Спецификации должны содержать данные о поставщике, методе производства, его месте расположения и видах животных, из которых извлекаются эти исходные материалы, а также способы их контроля. Особенную роль играют микробиологические методы контроля.
- Результаты испытаний исходных материалов должны соответствовать спецификациям. Если проведение испытаний занимает много времени (например, яйца (эмбрионы) безлейкозных линий птиц), может возникнуть необходимость обработки исходных материалов до получения результатов аналитических испытаний. В этих случаях получение разрешения на реализацию готовой продукции зависит от положительных результатов испытания исходных материалов.
- Особое внимание следует уделять данным о системе обеспечения качества, принятой у поставщика, об оценке пригодности источников материалов и требуемых для обеспечения качества тестах.
- Там, где это возможно, следует стерилизовать исходные материалы тепловым методом. Допускается использовать и другие аттестованные методы, например, ионизирующее излучение.
Среды
- Способность сред поддерживать необходимый рост клеток должна быть заранее подтверждена соответствующим образом.
- Предпочтительно, чтобы среды стерилизовались на месте (in situ) или на линии. Предпочтительным является метод тепловой обработки. Газы, среды, кислоты, щелочи, пеногасители и другие вещества, вводимые в стерильный биореактор, должны быть стерильными.
Система посевных материалов и банков клеток
- Для предотвращения нежелательного изменения свойств посевного материала вследствие многократных пересевов или большого числа генераций, производство иммунных лекарственных средств для животных, получаемых из микробных, клеточных или тканевых культур, или размножением в эмбрионах и животных, должно быть основано на системе посевных материалов или банков клеток.
- Количество генераций (удвоений, пассажей) между посевным материалом или банком клеток и готовой продукцией должно соответствовать данным нормативной документации и требованиям, установленным при регистрации лекарственного средства.
- Посевные материалы и банки клеток следует соответствующим образом характеризовать и контролировать на наличие загрязнений. Посевные материалы и банки клеток должны создаваться, содержаться и использоваться таким образом, чтобы снизить до минимума риск загрязнения или любого изменения. При создании посевных материалов или банков клеток не допускается одновременная работа в той же зоне или тем же персоналом с другими живыми или инфекционными материалами (например, вирусами или клеточными линиями).
- Для защиты посевного материала и банка клеток, а также работающего с ними персонала и внешней среды посевные материалы и банк клеток должны создаваться с соблюдением установленных требований к окружающей среде.
- Происхождение, форма и условия хранения посевного материала должны быть полностью описаны в соответствующей документации. Необходимо иметь доказательства стабильности и воспроизводимости посевных материалов и клеток. Емкости для хранения должны быть герметично закрыты, четко маркированы и храниться при требуемой температуре. Условия хранения должны контролироваться. Необходимо вести тщательный учет каждой единицы хранения.
- К работе с материалом допускаются только специально назначенные сотрудники. Работа должна проводиться под контролем ответственного лица. Различные посевные материалы и банки клеток следует хранить таким образом, чтобы не допускать перепутывания или перекрестного загрязнения. Желательно разделять посевные материалы и банки клеток на части и хранить их в различных местах, чтобы уменьшить опасность полной потери.
Принципы работы
- В ходе технологических процессов следует избегать или сводить к минимуму образование капель или пены. Процессы центрифугирования или смешивания, которые могут привести к образованию капель, должны проводиться в изолированных или чистых/изолированных зонах во избежание переноса живых организмов.
- Случайные проливы жидкостей, особенно содержащих живые организмы, необходимо быстро и безопасным способом ликвидировать. Для каждого типа микроорганизмов следует использовать аттестованные методы деконтаминации. При использовании различных штаммов бактерий одного вида или очень похожих вирусов этот процесс может быть аттестован только для одного из них, если данные о существенном различии их устойчивости к воздействию используемого агента отсутствуют.
- Операции, включающие в себя перемещение таких материалов, как стерильные среды, культуры или продукты, должны проводиться в предварительно стерилизованных закрытых системах. Если это невозможно, операции по перемещению материалов должны проводиться в однонаправленном (ламинарном) потоке воздуха.
- Добавление сред или культур в биореакторы (ферментаторы) и другие сосуды должно проводиться в тщательно контролируемых условиях, обеспечивающих невозможность внесения загрязнения. При добавлении культур необходимо тщательно проверять правильность соединения сосудов.
- При необходимости (например, когда два и более ферментатора расположены в одной зоне), отверстия для отбора проб и внесения добавок должны стерилизоваться паром (после подсоединения, перед подачей продукта и перед отсоединением). В других случаях допускается химическая дезинфекция входных отверстий и защита соединений под ламинарным потоком воздуха.
- Оборудование, лабораторная посуда, внешние поверхности упаковок с продукцией и другие подобные материалы перед перемещением из изолированной зоны должны быть дезинфицированы с использованием аттестованного метода (пункт 47 данного приложения). Особую проблему может вызывать ведение документации на серию продукции. Только абсолютный минимум документации, необходимой для соблюдения требований настоящего стандарта, должен поступать в зону и покидать ее. При случайном загрязнении (например, каплями или аэрозолями) или при использовании экзотических микроорганизмов бумажная документация должна дезинфицироваться в шлюзе для перемещения оборудования или передаваться с использованием фотокопии или факса.
- Жидкие или твердые отходы, такие как дебрис после отбора материала из яиц, одноразовые бутыли для культур, нежелательные культуры или биологические агенты, целесообразно стерилизовать или дезинфицировать перед их удалением из изолированной зоны. В некоторых случаях приемлемыми считаются и другие методы, например использование герметичных контейнеров или трубопроводов.
- Необходимо тщательно контролировать, чтобы в производственную зону попадали только те предметы и материалы, в т. ч. документация, которые относятся к производимой продукции. Для контроля за соответствием количества вносимых и выносимых предметов и материалов во избежание их накопления в производственном помещении должна действовать система учета.
- Термостойкие предметы и материалы должны поступать в чистую или чистую изолированную зону через проходные автоклавы или печи. Чувствительные к нагреву предметы и материалы должны поступать через воздушный шлюз с блокируемыми дверями, где эти предметы и материалы подвергаются дезинфекции. Допускается стерилизация предметов и материалов в другом месте, если они поступают через шлюз в двойной оболочке с соблюдением необходимых мер предосторожности.
- Во избежание загрязнения или подмены культуры клеток или микроорганизмов в инкубационный период необходимо принимать меры предосторожности. Должна существовать методика очистки и дезинфекции инкубаторов. Упаковки, находящиеся в инкубаторах, должны быть четко и тщательно маркированы.
- В одном помещении допускается одновременное проведение операций только с одним биологическим агентом, за исключением операций по смешиванию и последующему наполнению или при использовании полностью закрытых систем. В промежутках между операциями с различными живыми биологическими агентами производственные помещения должны проходить эффективную дезинфекцию.
- Продукты должны инактивироваться путем добавления инактиватора с последующим тщательным перемешиванием. После этого смесь должна переноситься во второй стерильный сосуд, за исключением случаев, когда форма и размер сосуда позволяют его легко переворачивать и встряхивать таким образом, чтобы все внутренние поверхности смачивались конечной смесью культураинактиватор.
- Сосуды, содержащие инактивированный продукт, не допускается открывать. Из этих сосудов не допускается отбирать пробы в зонах, где содержатся живые биологические агенты. Всю последующую обработку инактивированных продуктов следует проводить в чистых зонах А и В или в закрытом оборудовании, предназначенном для инактивированных продуктов.
- Необходимо уделять серьезное внимание аттестации (испытаниям) методов стерилизации, дезинфекции, удаления вирусов и инактивации.
- Наполнение, по возможности, следует проводить немедленно после завершения производственных операций. До начала операции наполнения емкости с нерасфасованной продукцией должны быть герметично закрыты, маркированы и храниться при установленных температурных условиях.
- Должна существовать система, обеспечивающая контроль целостности и герметичности упаковок после наполнения.
- Флаконы, содержащие живые биологические агенты, должны быть закрыты крышками таким образом, чтобы обеспечивалась невозможность загрязнения другой продукции или проникание живых агентов в другие зоны или во внешнюю среду.
- Между наполнением первичных упаковок, их маркировкой и упаковкой по различным причинам может пройти определенный отрезок времени. Должны быть разработаны и документально оформлены методики, описывающие порядок хранения немаркированных контейнеров, обеспечивающие невозможность их подмены и надлежащие условия хранения. Особое внимание следует уделять хранению термои светочувствительной продукции. Температура хранения должна быть задана.
- На каждой технологической стадии следует проводить сопоставление реального и ожидаемого выхода продукции. Все существенные отклонения должны быть расследованы.
Контроль качества
- В обеспечении стабильности качества биологических лекарственных средств особое значение имеет внутрипроизводственный контроль. Виды контроля, которые имеют решающее значение для оценки качества (например, контроль на отсутствие вирусов), но не могут быть проведены на готовой продукции, должны выполняться на одной из предшествующих стадий производства.
- Для повторения или подтверждения результатов контроля качества серии продукции может возникнуть необходимость хранения при соответствующих условиях достаточного объема образцов промежуточных продуктов.
- Может возникнуть необходимость непрерывного контроля параметров в ходе процесса производства (например, непрерывного контроля физических параметров в ходе ферментации).
- Распространенной практикой является непрерывное культивирование биологической продукции. При таком методе производства необходимо принять во внимание особые требования к организации контроля качества продукции.
Производство добавок к лекарственным кормам
В настоящем приложении использованы следующие термины: лекарственные корма (medicated feeding stuff): Любая смесь лекарственных средств для животных и кормов, выпускаемая в готовом виде, предназначенная для кормления животных без дальнейшей обработки, которая рассматривается как лекарственное средство благодаря своим лечебным или профилактическим свойствам. добавки к лекарственным кормам (премиксы) (pre-mix): Любые лекарственные средства для животных, приготовленные заранее для последующего использования в производстве лекарственных кормов.- Производство добавок к лекарственным кормам требует использования большого количества растительных материалов, привлекающих насекомых и грызунов. Помещения для производства добавок следует проектировать, оборудовать и эксплуатировать таким образом, чтобы свести к минимуму опасность проникания насекомых и грызунов (часть I, 3.4). С целью борьбы с ними следует проводить регулярную обработку помещений.
- Из-за большого объема пыли, образующегося при производстве нерасфасованных веществ для добавок, особое внимание необходимо уделять мерам по облегчению очистки помещений и предотвращению перекрестного загрязнения (часть I, 3.14), например, применением герметичных транспортеров и пылепоглотителей. Наряду с использованием такого оборудования следует проводить тщательную и регулярную очистку производственных помещений.
- Следует обеспечить единообразие выполнения (от серии к серии) стадий технологического процесса, которые оказывают существенное отрицательное влияние на стабильность активных веществ (например, использование пара при производстве гранул).
- Производство добавок целесообразно организовывать в специализированных зонах, вынесенных за пределы основных производственных помещений. В противном случае эти специализированные помещения следует окружить буферными зонами, чтобы опасность загрязнения других производственных зон была минимальной.
Производство препаратов против эктопаразитов
- Несмотря на ограничение, указанное в части I, 3.6 настоящего стандарта, препараты против эктопаразитов, предназначенные для наружного применения, относящиеся к лекарственным средствам для животных и включенные в лицензию на производство, могут производиться и наполняться в зонах, предназначенных для изготовления пестицидов, по принципу разделенных во времени циклов производства. Такие зоны не предназначены для производства других видов лекарственных средств для животных.
- Для предотвращения перекрестного загрязнения следует использовать аттестованные методы очистки. Следует принять меры по обеспечению безопасного хранения препаратов для животных в соответствии с требованиями настоящего стандарта.
Производство лекарственных средств для животных, содержащих пенициллины
- Использование пенициллинов в ветеринарии не представляет такого риска с точки зрения гиперсенсибилизации животных, как в случае их использования человеком. Зарегистрированные случаи гиперсенсибилизации у лошадей и собак были обусловлены другими токсичными веществами (например, ионофорными антибиотиками для лошадей). Такие препараты рекомендуется производить в специально предназначенных изолированных помещениях (часть I, 3.6), но эта рекомендация может не распространяться на случаи, когда помещения предназначены только для производства лекарственных средств для животных. Однако необходимо принять все необходимые меры по предотвращению перекрестного загрязнения и обеспечению безопасности персонала в соответствии с требованиями настоящего стандарта. При использовании общих помещений производство продукции, содержащей пенициллины, должно быть организовано по принципам разделенных во времени циклов производства и должно сопровождаться аттестованными методиками деконтаминации и очистки.
- В связи с большими объемами окончательных упаковок некоторых лекарственных средств для животных (в частности, добавок) производителю может оказаться неудобным хранить образцы каждой серии продукции в их окончательной упаковке. Однако производитель должен обеспечить хранение достаточного количества архивных образцов каждой серии продукции в соответствии с требованиями настоящего стандарта.
- В любых случаях упаковка для хранения архивных образцов должна быть изготовлена из того же материала, что и маркированная первичная упаковка, в которой этот продукт реализуется на рынке.
Стерильные лекарственные средства для животных
Лекарственные средства для животных, подлежащие финишной стерилизации, могут производиться в чистых зонах более низкого класса, чем это требуется в приложении 1 (если решение об этом принято в установленном порядке), однако тип зоны помещений должен быть не ниже D.Общие положения
Производство радиофармацевтических препаратов должно быть организовано в соответствии с Правилами производства и контроля качества лекарственных средств GMP (части I и II настоящего стандарта). Данное приложение устанавливает требования, специфические для производства радиофармацевтических препаратов. Примечания:- Данное приложение не распространятся на приготовление радиофармацевтических препаратов в специализированных аптеках (в лечебных учреждениях или действующих самостоятельно), с использованием радионуклидных генераторов и наборов реагентов в соответствии с лицензией (регистрационными документами).
- В соответствии с требованиями радиационной безопасности ответственность за применение радиации в медицинских целях лежит на лечебных учреждениях (врачах). При применении радиофармацевтических препаратов в диагностических и терапевтических целях должна быть обеспечена возможность участия специалиста по медицинской физике и выполнены другие требования.
- Данное приложение распространяется также на радиофармацевтические препараты, использующиеся в клинических испытаниях.
- Транспортирование радиофармацевтических препаратов выполняется в соответствии с требованиями по радиационной безопасности Международного Агентства по атомной энергии (МАГАТЭ) и национальными требованиями.
- Могут использоваться методы, отличающиеся от приведенных в данном приложении, но позволяющие выполнить требования по обеспечению качества продукции. При применении этих методов следует показать, что они обеспечивают уровень качества, эквивалентный, по крайней мере, требованиям данного приложения.
Введение
- Производство радиофармацевтических препаратов и обращение с ними представляет потенциальную опасность. Уровень риска зависит от типа излучения, энергии излучения и периода полураспада радионуклидов. Особое внимание следует уделить предотвращению перекрестного загрязнения, наличию остатков радиоактивных материалов и удалению отходов.
- Из-за того что радионуклиды имеют короткий срок хранения, допускается выпускать некоторые радиофармацевтические препараты до завершения контроля качества. В этом случае в специальной инструкции должен быть четко и подробно определен порядок выпуска препаратов, включая ответственность персонала и требования к непрерывной оценке эффективности системы обеспечения качества.
- Областью применения данного приложения является деятельность промышленных предприятий, ядерных центров, институтов и ПЭТ-центров по производству и контролю качества:
- радиофармацевтических препаратов;
- радиофармацевтических препаратов для ПЭТ (позитронно-эмиссионной томографии);
- радиоактивных предшественников для производства радиофармацевтических препаратов;
- радионуклидных генераторов.
| Вид производства | Правила GMP не распространяются* | Следует выполнять требования настоящего стандарта. Части II и I (по мере приближения стадии производства к готовому продукту и соответствующие приложения) | |||
| Радиофармацевтические препараты Радиофармацевтические препараты для ПЭТ Радиоактивные предшественники | Продукция реакторов и циклотронов | Химический синтез | Стадии очистки | Переработка, формирование, упаковывание (наполнение, фасование) | Асептическое производство или финишная стерилизация |
| Радионуклидные генераторы | Продукция реакторов и циклотронов | Технологический процесс | |||
| * Мишень и система передачи от циклотрона к установке синтеза могут рассматриваться как первая стадия производства активных фармацевтических субстанций. | |||||
- Производитель готового радиофармацевтического препарата должен иметь описание технологического процесса производства активной фармацевтической субстанции, готового лекарственного средства и указать, какие требования GMP (часть I или II настоящего стандарта) распространяются на различные технологические операции/стадии.
- Производство радиофармацевтических препаратов должно выполняться в соответствии с требованиями норм радиационной безопасности.
- Производство радиофармацевтических препаратов, предназначенных для парентерального
- Требования к радиофармацевтическим препаратам и методам их контроля установлены в фармакопейных статьях или других документах*.
Клинические испытания
- На производство радиофармацевтических препаратов, предназначенных для клинических исследований, распространяются также требования приложения 13 к настоящему стандарту.
Обеспечение качества
- Обеспечение качества при производстве радиофармацевтических препаратов имеет особое значение ввиду их специфических особенностей, малых объемов серий и в некоторых случаях необходимости применения до завершения операций по контролю качества.
- Защита продукции от загрязнений и перекрестных загрязнений должна быть обеспечена так же, как и при производстве любых лекарственных средств. Но в данном случае предъявляется дополнительное требование по защите персонала от ионизирующего излучения. В этих условиях система обеспечения качества приобретает исключительно важное значение.
- Следует проводить оценку и регистрацию данных по контролю помещений и оборудования в соответствии с их значением в технологическом процессе.
- При производстве радиофармацевтических препаратов следует в необходимом объеме проводить аттестации (испытания) с учетом анализа рисков, который позволяет оценить объем работ по аттестации (испытаниям) в соответствии с требованиями данного стандарта и нормативных документов по радиационной безопасности.
Персонал
- Все технологические операции должны выполняться персоналом, имеющим специальную подготовку по радиационной безопасности. Персонал, занятый в производстве, контроле качества и выпуске радиофармацевтических препаратов, должен пройти специальное обучение, связанное с особенностями обеспечения качества этих препаратов. Уполномоченное лицо несет полную ответственность за выпуск радиофармацевтических препаратов.
- Весь персонал, работающий в зонах производства радиофармацевтических препаратов (включая занятый уборкой и техническим обслуживанием), должен проходить дополнительное обучение, связанное со спецификой процессов и продукции.
- Если производственные помещения и оборудование используются также для проведения исследований, то исследовательский персонал должен пройти обучение по правилам GMP. Служба обеспечения качества должна рассматривать и давать разрешение на проведение работ, связанных с исследованиями, для того чтобы исключить их опасное влияние на производство.
Помещения и оборудование
Общие положения
- Производство радиофармацевтических препаратов должно быть организовано в контролируемых зонах, в которых выполняются требования к окружающей среде и радиационной безопасности. Все технологические операции должны выполняться в специальных помещениях и на специальном оборудовании, предназначенном для производства радиофармацевтических препаратов.
- Следует принять меры по предотвращению перекрестных загрязнений от персонала, материалов, радиоактивных материалов и пр. Везде, где это возможно, следует использовать закрытое или изолированное оборудование. При использовании открытого оборудования следует принять меры по сведению риска загрязнений к минимуму. При оценке риска следует показать, что чистота окружающей среды удовлетворяет требованиям, предъявляемым к типу выпускаемой продукции.
- Вход в производственные зоны должен осуществляться через комнаты переодевания (санпропускники) и должен быть ограничен только для персонала, имеющего право доступа в них.
- Следует проводить контроль рабочих мест и окружающей среды на наличие (концентрацию) радиоактивных загрязнений, частиц и микроорганизмов. Порядок проведения контроля устанавливается при аттестации в эксплуатируемом состоянии.
- Для обеспечения надлежащей работы помещений и оборудования следует организовать их техническое обслуживание, проводить калибровку (поверку) и аттестацию (испытания). Эти работы должны выполняться подготовленным персоналом, а факт их проведения и полученные результаты должны оформляться документально.
- Следует принять меры по защите от радиоактивного загрязнения помещений и оборудования. Следует организовать контроль радиоактивных загрязнений непосредственно с помощью дозиметров или косвенно методом мазков.
- Поверхности оборудования, соприкасающиеся с продуктом, не должны вступать с ним в реакцию, ничего не привносить в него и не абсорбировать продукт, чтобы не изменить его свойства.
- Рециркуляция воздуха из помещений, в которых выполняется работа с радиофармацевтическими препаратами, не допускается за исключением случаев, когда применение рециркуляции обосновано. В вытяжных системах должна быть предусмотрена защита от загрязнения окружающей среды радиоактивными частицами и газами. В контролируемых зонах должна быть предусмотрена защита от загрязнения частицами и микроорганизмами.
- Чтобы не допустить распространения радиоактивных частиц, может оказаться необходимым поддерживать отрицательное давление в зонах, где находится открытый продукт. В то же время следует защитить продукт от загрязнений из окружающей среды. Это может быть достигнуто за счет применения барьерной технологии и воздушных шлюзов, служащих для поддержания перепада давления.
Производство стерильной продукции
- Стерильные радиофармацевтические препараты разделяются на две группы:
- препараты, производство которых должно быть организовано в асептических условиях.
- препараты, подлежащие финишной стерилизации.
- Для определения требований к перепадам давления, направлению потока воздуха и его качества могут использоваться методы анализа рисков.
- В закрытых и автоматизированных системах, представляющих собой, как правило, «горячие камеры» с размещением в них установок химического синтеза, систем очистки, стерилизующей фильтрации «на линии», должна быть предусмотрена зона С. В горячие камеры, находящиеся в закрытом состоянии, должен подаваться воздух с высокой степенью чистоты. Асептические операции должны выполняться в зоне А.
- До начала производства сборка стерильного оборудования и компонентов (трубок, стерилизующих фильтров), укупорка и герметизация стерильных флаконов должны выполняться в асептических условиях.
Документация
- Порядок разработки, пересмотра, утверждения и выдачи документов, относящихся к производству радиофармацевтических препаратов, должен быть изложен в специальной инструкции.
- Требования к исходным и упаковочным материалам, этикеткам и другим средствам маркировки, критическим промежуточным материалам и готовой продукции должны быть указаны в спецификациях. Спецификации также разрабатываются на критические материалы и компоненты (вспомогательные вещества, уплотнения, наборы для стерилизующей фильтрации и др.), используемые в процессе и способные оказать критическое влияние на качество продукции.
- Следует установить допустимые предельные значения изменений характеристик радиофармацевтических препаратов, включая требования к выпуску и сроку хранения (например, химической идентичности изотопов, объемной активности, радионуклидной чистоты и удельной активности).
- В протоколах на использование, очистку, дезактивацию, дезинфекцию (стерилизацию), техническое обслуживание следует указывать дату и время выполнения операции, заверять протокол подписью лица, выполнившего работу, и, при необходимости, указывать наименование продукта и номер серии.
- Протоколы следует хранить не менее чем в течение трех лет, если иное не указано в других документах.
Производство
- Одновременное производство различных радиофармацевтических препаратов в одной рабочей зоне (горячей камере, ламинарной зоне или шкафу) не допускается, что вызвано необходимостью снижения до минимума риска перекрестного загрязнения радиоактивными веществами или перепутывания материалов.
- Процессы и оборудование подлежат аттестации (испытаниям), в т. ч. системы с компьютерным управлением, в соответствии с приложением 11 к настоящему стандарту. Новые процессы подлежат перспективной аттестации.
- Критические параметры следует, как правило, определить до проведения аттестации или в ее процессе. При этом следует определить допустимые предельные значения изменений параметров, необходимые для стабильного производства.
- Для продуктов, наполняемых в асептических условиях, следует проводить контроль целостности мембранных фильтров, принимая во внимание необходимость обеспечения радиационной безопасности и сохранения стерильности фильтров.
- Учитывая радиационную активность готового продукта, допускается наносить маркировку на первичную упаковку до начала производства. На стерильные пустые закрытые флаконы может быть нанесена маркировка с частичной информацией до операции наполнения, при этом стерильность не должна быть нарушена и не должно быть помех для визуального контроля наполненных флаконов.
Контроль качества
- Некоторые радиофармацевтические препараты могут быть выпущены и использованы на основе оценки документации на серию до завершения всех химических и микробиологических испытаний.
- b) получение разрешения на выпуск от уполномоченного лица после проведения оценки окончательных результатов аналитического контроля, всех отклонений от нормального процесса, которые должны быть оформлены документально, обоснованы и утверждены. Если некоторые результаты контроля невозможно получить до использования препарата, то уполномоченному лицу следует оформить разрешение на выпуск препарата условно до его использования и окончательно оформить разрешение на выпуск препарата после получения всех результатов контроля.
- Большинство радиофармацевтических препаратов используются в течение короткого периода времени. Срок годности препарата должен быть четко указан.
- Препараты, содержащие радиоактивные изотопы с большим периодом полураспада, следует контролировать на соответствие всем требованиям до оформления разрешения на выпуск уполномоченным лицом.
- Пробы могут храниться до проведения контроля в течение некоторого времени, чтобы радиоактивность была снижена до допустимого уровня. Все виды контроля, включая контроль на стерильность, должны быть проведены как можно быстрее.
- Порядок оценки продукции и результатов контроля, которую следует провести до выпуска продукции, должен быть изложен в специальной инструкции.
- Продукция, не соответствующая установленным требованиям, должна быть отклонена. Если предусмотрена переработка материала, то она должна выполняться по инструкции. Готовая продукция должна соответствовать установленным требованиям, что должно быть подтверждено до ее выпуска. Не допускается переработка возвращенной продукции, с которой следует обращаться как с радиоактивными отходами.
- В специальной инструкции должен быть определен порядок действий уполномоченного лица в случае обнаружения несоответствия продукции требованиям спецификации после ее отгрузки до истечения срока годности. Такие случаи должны быть расследованы с разработкой плана мероприятий по их предупреждению и документальному оформлению.
- При необходимости следует информировать ответственный персонал лечебного учреждения. Это следует выполнять в порядке, обеспечивающем прослеживаемость всех действий.
- Должен быть разработан порядок контроля исходных материалов. При выборе и утверждении поставщика следует убедиться в том, что поставляемые им материалы неизменно соответствуют требованиям спецификаций. Исходные и упаковочные материалы и вспомогательные материалы для критических процессов должны приобретаться только у утвержденных поставщиков.
Контрольные и архивные образцы
- Из каждой серии нерасфасованной продукции должны быть отобраны пробы (контрольные и архивные образцы), которые должны храниться не менее шести месяцев после истечения срока годности готовой продукции, если иное не установлено при проведении анализа рисков.
- Пробы исходных материалов, за исключением растворителей, газов и воды, используемой в производстве, должны храниться не менее двух лет после выпуска продукции. Этот срок может быть сокращен, если в спецификации на материал указан более короткий период стабильности.
- Соглашением с компетентными органами может быть определен другой порядок отбора проб (контрольных и архивных образцов) исходных материалов, продукции, изготовленной по индивидуальному заказу или в малых количествах, или если их хранение может вызвать особые трудности.
Реализация
- Допускается реализация готовой продукции до получения результатов испытаний, если сам процесс реализации находится под контролем и препарат не будет использован до получения результатов испытаний и их оценки ответственным лицом.
Термины и определения
горячая камера (Hot-cell): Защищенное (экранированное) рабочее место для производства и обращения с радиоактивными материалами. Горячая камера не обязательно должна быть изолятором. приготовление (preparation): Приготовление радиомеченного набора радионуклидом, эллюированным из генераторов или радиоактивными предшественниками в лечебном учреждении. Наборы, генераторы и радиоактивные предшественники должны быть зарегистрированы в установленном порядке, а предприятия-производители должны иметь лицензию на выпуск данной продукции. производство (manufacturing): Изготовление, контроль качества, выпуск и доставка радиофармацевтических препаратов, полученных из активной субстанции и других исходных материалов. уполномоченное лицо (Qualified Person): См. приложение 16 к настоящему стандарту.Область применения
Методы производства биологических активных субстанций и биологических лекарственных средств для человека («биологических активных субстанций и лекарственных средств») являются критическим фактором в выполнении установленных требований. Свойства биологических активных субстанций и лекарственные средства в значительной степени зависят от методов производства. Данное приложение дает руководство для всех активных субстанций и лекарственных средств, отнесенных к биологическим, за исключением «Новейших терапевтических лекарственных средств – НТЛС» (Advanced Therapy Medicinal Products – ATMP), которые рассматриваются в части IV правил GMP. Данное приложение и состоит двухосновных частей:- Часть А содержит дополнительное руководство по производству биологических активных субстанций и лекарственных средств, начиная от посевного материала и банков клеток до завершающих операций и контроля;
- Часть В содержит дальнейшее руководство для отдельных типов биологических активных субстанций и лекарственных средств.
- Стадия производства биологических активных субстанций до точки, когда они являются стерильными – первичным руководством является часть II настоящего стандарта;
- Вид продукта. Данное приложение относится ко всем лекарственным средствам, определенным как биологические, за исключением НТЛС (ATMP).
| Вид и источник материла | Пример продукции | Применение данного руководства к технологическим стадиям выделено серым фоном | |||
| 1. Животное или растение, не трансгенное | Гепарины, инсулины, энзимы, белки, экстракты аллергенов, иммунная сыворотка | Сбор растений, орган, материал животного происхождения или жидкость1 | Резка (измельчение), смешивание и/или первичная переработка | Изоляция и очистка | Приготовление, наполнение |
| 2. Вирус или бактерии /ферментация/ культура клеток | Вирусная или бактериальная вакцина; энзимы, белки | Определение и поддержание MCB2, WCB, MVS, WVS | Культура клеток и/или ферментация | Инактивация, если требуется, изоляция и очистка | Приготовление, наполнение |
| 3. Биотехнология – ферментация/ культура клеток | Рекомбинантные продукты, Mab, аллергены, вакцины | Определение и поддержание MCB и WCB, MSL, WSL | Культура клеток и/или ферментация | Изоляция, очистка, модификация | Приготовление, наполнение |
| 4. Животное трансгенное | Рекомбинантные белки | Основной и рабочий трансгенные банки | Сбор, резка, смешивание и/или первичная переработка | Изоляция, очистка, модификация | Приготовление, наполнение |
| 5. Растение трансгенное | Рекомбинантные белки, вакцина, аллергены | Основной и рабочий трансгенные банки | Выращивание, сбор3 | Первичная экстракция, изоляция, очистка, модификация | Приготовление, наполнение |
| 6. Человек | Энзимы, полученные из мочи, гормоны | Сбор жидкости4 | Смешивание и/или первичная переработка | Изоляция и очистка | Приготовление, наполнение |
| 7. Человек | Продукты из клеток тканей | Создание о поддержание главного и рабочего банков клеток, главного и рабочего вирусных посевных материалов5 | Первичная переработка, изоляция и очистка | Изоляция клеток, культур, очистка, комбинирование с неклеточными компонентами | Приготовление, комбинирование, наполнение |
Основные принципы
Производство биологических активных субстанций и лекарственных средств имеет свою специфику, определяемую характером продукции и технологией производства. При производстве, контроле и введении биологических лекарственных средств необходимо принимать специальные меры предосторожности. В отличие от обычных лекарственных средств, которые производятся с использованием химических и физических методов, обеспечивающих высокую степень стабильности, производство биологических активных субстанций и лекарственных средств связано с биологическими процессами и материалами, такими как культивирование клеток или экстракция материала из живых организмов. Эти биологические процессы характеризуются вариабельностью, что приводит к непостоянству спектра и природы сопутствующих продуктов. В результате для этого вида материалов особенно важными являются методы анализа рисков, которые должны использоваться при разработке стратегии (порядка) контроля на всех стадиях производства для сведения вариабельности к минимуму и снижения возможности загрязнений и перекрестных загрязнений. 1 Пояснения к области применения принципов GMP дано в приложении В1. 2 Пояснения к области применения принципов GMP дано в разделе «посевные материалы и банки клеток». 3 Руководство HMPC «Правила выращивания и сбора» (Good Agricultural and Collection Practice» – ЕМЕА/HHMPC/246816/2005. 4 Пояснения к области применения принципов GMP дано в разделе «Область применения». 5 Клетки и ткани человека должны соответствовать Директиве 2004/23/ЕС и последующим Директивам для этих стадий. Поскольку материалы, используемые в процессах культивирования, и условия их проведения сами предназначены для обеспечения роста специфических клеток и микроорганизмов, они могут способствовать росту посторонних микроорганизмов. Кроме того, у некоторых продуктов способность выдерживать широкий спектр методов очистки может быть ограниченной, особенно у тех, которые предназначены для инактивации и удаления случайного вирусного загрязнения. Основными факторами для сведения к минимуму таких загрязнений является разработка процессов, оборудования, помещений, инженерных систем, условия приготовления и добавления буферов и реагентов, отбора проб и обучения операторов. Требования к продукции (например, по фармакопейным статьям, регистрационному досье и разрешения на проведение клинических испытаний) должны устанавливать, могут ли вообще, а если могут, то до какой стадии субстанции и материалы иметь определенный уровень биозагрязнений или они должны быть стерильными. Производство также должно соответствовать другим требованиям регистрационного досье или разрешения на проведение клинических испытаний, например, к числу генераций (удвоений, пассажей) между посевными материалами или банками клеток. Работу с биологическими материалами, которые не подлежат стерилизации (например, фильтрованием) следует выполнять в асептических условиях, что позволят уменьшить свести риск внесения загрязнения до минимума. В этих случаях для аттестации специальных технологических методов, например, по удалению и инактивации вирусов, следует использовать действующие нормативные документы. Применение методов контроля параметров окружающей среды и, где требуется, систем очистки и стерилизации на месте совместно с применением закрытых систем могут существенно снизить риск случайных загрязнений и перекрестных загрязнений. Контроль обычно включает методы биологического анализа, которые обычно имеют большую вариабельность по сравнению с физико-химическими методами. Поэтому для устойчивости технологического процесса при производстве биологических активных субстанций и лекарственных средств особое значение имеют методы внутрипроизводственного контроля. Биологические лекарственные средства, в состав которых входят ткани или клетки человека должны соответствовать действующему законодательству в отношении требований к прослеживаемости. Должно быть дано уведомление о серьезных отрицательных реакциях и событиях и установлены требования к кодированию, производству, консервированию, хранению и реализации тканей и клеток человека, сбору и контролю, которые должны соответствовать системой качества согласно данному приложению. Биологические активные субстанции и лекарственные средства должны соответствовать действующему законодательству об уменьшения риска передачи возбудителя губчатой энцефалопатии животных и латентных вирусов через лекарственные средства для медицинского применения и для применения в ветеринарии.Часть А. Общее руководство
Персонал
- Все сотрудники, работающие в зонах производства биологических активных субстанций и продукции (в том числе персонал, занятый очисткой, обслуживанием или контролем качества), должны проходить первоначальное и дополнительное обучение в соответствии с их обязанностями и особенностями производимой продукции, включая специальные меры по безопасности для защиты продукта, персонала и окружающей среды.
- Для обеспечения безопасности продукции следует учитывать состояние здоровья персонала. При необходимости персонал, занятый в производстве, техническом обслуживании, контроле и уходе за животными (в том числе контролеры) должен проходить вакцинацию специальными вакцинами и регулярное медицинское обследование.
- Персонал не допускается к работе в производственной зоне при любых изменениях в состоянии здоровья, которые могут оказать отрицательное влияние на качество продукции, с соответствующим документальным оформлением. К производству вакцины БЦЖ и препаратов туберкулина допускаются только сотрудники, которые регулярно проходят проверки иммунного статуса или рентгеновское обследование грудной клетки. Контроль здоровья персонала следует организовать с учетом фактора риска. Персоналу, связанному с опасными операциями, должны оказываться медицинские консультации.
- При необходимости сведения к минимуму возможности перекрестных загрязнений следует ограничить передвижения всего персонала (включая работников, занятых контролем качества, техническим обслуживанием и уборкой) на основе принципов анализа рисков. Как правило, не допускается переход сотрудников из зон, где возможен контакт с живыми микроорганизмами, генетически модифицированными организмами, токсинами или животными, в зоны, где проводятся работы с другой продукцией, инактивированными продуктами или различными организмами. Если подобных переходов избежать невозможно, следует принять меры на основе принципов анализа рисков*.
Помещения и оборудование
- Требования к чистоте производственных помещений по частицам и микроорганизмам являются частью стратегии контроля и зависят от вида активных субстанций, промежуточной и готовой продукции и технологической стадии с учетом уровня загрязнения исходных материалов и риска загрязнения готовой продукции. В программу текущего контроля следует включать методы обнаружения специфических микроорганизмов (например, организмов-хозяинов, дрожжей, плесеней, анаэробов и т. д.), определенных в соответствии с принципами анализа рисков.
- Классификация помещений для производства и хранения, процессов и окружающей среды, должны предусматривать защиту продукции от внешних загрязнений. Предупреждение загрязнений является более предпочтительным по сравнению с обнаружением и удалением, не смотря на то, что загрязнение может быть скорее обнаружено в культуре клеток и в ходе таких процессов как ферментация. Для процессов, не являющихся закрытыми, когда продукт открыт непосредственно в окружающую среду (например, при загрузке добавок, питательной среды, буферов, газов) может быть организован контроль на месте, включая инженерный контроль и контроль окружающей среды на основе принципов анализа рисков. При этом следует принимать во внимание соответствующие разделы приложения 1 к настоящему стандарту1 (правилам GMP), определяя каскады перепадов давления и связанные с ними методы контроля.
- Для работы с клетками, устойчивыми к технологической среде, следует предусматривать отдельные производственные зоны. Такие выделенные зоны требуются в производствах с патогенными организмами (уровни безопасности 1 и 2 по классификации РФ).
- Многономенклатурное производство допускается при выполнении следующих или эквивалентных условий (в зависимости от вида продукции) и принятии мер в составе эффективной стратегии предупреждения перекрестных загрязнений:
- Знание основных характеристик всех клеток, организмов и любых других факторов (например, патогенности, возможности обнаружения, живучести, чувствительности к инактивации) в пределах того же производства;
- Производство многих малых серий и при различии характеристик исходных материалов. Например, в ходе разработки стратегии контроля следует учитывать состояние здоровья доноров и риск полной потери продукции при оценке возможности одновременной работы;
- Для предотвращения проникания живых организмов и спор в не относящиеся к работе с ними зоны или оборудование, следует определить все возможные пути перекрестных загрязнений и использовать одноразовые компоненты и инженерные решения, например, закрытые системы;
- Следует удалять организмы и споры до начала последующего производства других продуктов. Эти меры также относятся средства отопления, вентиляции и кондиционирования воздуха;
- Контроль окружающей среды с учетом специфики получаемых микроорганизмов выполняется в соседних зонах в ходе производства и после завершения очистки и дезинфекции, если микроорганизмы могут быть устойчивыми к технологической среде и есть доступные методы. Следует учесть риски, исходящие от контрольных приборов (например, сенсоров частиц) в зонах с обращением с живыми и/или спорообразующими организмами;
- Продукция, основное и вспомогательное оборудование (например, для калибровки и аттестации) и одноразовые материалы перемещаются и удаляются из таких зон с соблюдением мер предосторожности от загрязнения других зон, другой продукции и различных технологических стадий (например, с предотвращением загрязнения инактивированных продуктов или анатоксинов неинактивированными продуктами).
- Производство циклами.
- Необходимость в специализированном (выделенном) оборудовании для завершающих операций2 зависит от всех приведенных выше указанных условий и таких дополнительных факторов как специфические особенности биологических лекарственных средств и характеристики другой продукции, включая любые небиологические продукты, производимые в тех же помещениях. К другим методам контроля на завершающих стадиях могут относиться специфические последовательности добавления, скорость смешивание, контроль времени и температуры, пределы для воздействия света, изоляция и очистка при проливе.
- В прежней версии этого приложения в правилах GMP EC и в ГОСТ Р 52249–2009 формулировка была более точной: «Если подобных переходов избежать невозможно, персонал, занятый в таком производстве, должен строго выполнять требования по деконтаминации, в том числе к смене одежды и обуви, и, при необходимости, принимать душ». Замена ясных требований на общие фразы привела к утрате смысла (прим. разработчика стандарта).
- Методы и средства по изоляции (например, для охраны среды и безопасности оператора) не должны приводить к снижению качества продукции.
- Конструкция, монтаж и техническое обслуживание кондиционеров должны сводить к минимум риск перекрестного загрязнения между различными производственными зонами и могут, при необходимости, быть специализированными для отдельных зон. Решения по применению прямоточных систем следует принимать на основе анализа рисков.
- Для производства стерильной продукции следует использовать помещения с избыточным давлением. Для предотвращения распространения загрязнения за пределы специальных зон, подвергаемых воздействию патогенов, в этих зонах может быть предусмотрен отрицательный перепад давления.
- Конструкция оборудования, используемого для работы с живыми микроорганизмами и клетками, включая используемые для отбора проб, должна исключать возможность загрязнения этих культур из внешних источников в ходе технологического процесса.
- Конструкция первичной изоляции1 должна исключать риск утечки биологических агентов в непосредственно окружающую рабочую среду, что должно подтверждаться результатами периодических испытаний.
- Рекомендуется использовать системы «очистка на месте» (clean in place) и «стерилизация на месте» (sterilize in place). Конструкция вентилей на ферментаторах должна предусматривать возможность их стерилизации паром.
- Вентиляционные воздушные фильтры должны быть гидрофобными и аттестованы для эксплуатации в течение срока службы и подлежат контролю целостности через соответствующие интервалы времени на основе принципов анализа рисков.
- Жидкие отходы, которые могут содержать патогенные микроорганизмы, должны проходить эффективную нейтрализацию или дезинфекцию с целью снижения риска перекрестных загрязнений. Следует выполнять установленные требования по сведения к минимуму риска загрязнения внешней среды с учетом биологической опасности отходов.
- Вследствие вариабельности биологических продуктов или процессов следует отмеривать или взвешивать соответствующее/критическое сырье (например, питательные среды и буферы) в ходе технологического процесса. В этих случаях допускается хранение небольших запасов этого сырья в производственной зоне в течение установленного времени, определенного по таким критериям как длительность производства серии или производства циклами.
Животные
- В производстве биологических лекарственных средств используются разные виды животных, которые можно разделить на два большие группы:
- Живые животные, табуны, стада: примерами служат вакцины против полиомиелита (обезьяны), иммунные сыворотки против ядов змей и столбняка (лошади, овцы и козы), аллергены (кошки), вакцина против бешенства (кролики, мыши и хомяки), трансгенные препараты (козы, крупный рогатый скот);
- Материалы животного происхождения, полученные при вскрытии трупов или от таких предприятий как скотобойня; к примерам относятся энзимы, антикоагулянты и гормоны, получаемые от скотобоен (овцы и свиньи).
- Дополнительно к нормативным требованиям, касающимся трансмиссионной губчатой энцефалопатии крупного рогатого скота, следует контролировать другие опасные агенты (возбудители зоонозов; болезни, передаваемые животными) в соответствии с действующей программой и оформлять документально. К выполнению этой работы следует привлекать специалиста. Нужно расследовать случаи заболевания животных-доноров или животных, используемых в качестве сырья, в плане пригодности этих животных и пригодности животных, имевших контакт с больным животным, для постоянного использования (в производстве в качестве сырья и, для контроля качества и проведения испытаний), и принятые решения оформлять документально. Следует разработать порядок ретроспективного анализа, позволяющего принимать решение относительно годности биологической субстанции или лекарственного средства, в состав которых входит или при производстве которых использовался такой материал животного происхождения в качестве сырья или исходного материала. При принятии решения могут быть учтены результаты повторных испытаний или испытаний сохраняемых образцов от прежнего взятия материала от того же животного-донора (если применимо) для установления последнего донорства, которое дало отрицательный результат. Период выведения терапевтического средства, использовавшегося для лечения животного-донора или животногоисточника сырья, должен быть зарегистрирован и учитываться при принятии решении о выведении этого животного из программы на определенное время.
- Особое внимание следует уделять предотвращению и мониторингу инфекционных заболеваний у животных-доноров или животных-источников сырья.
- Для лекарственных средств, произведенных с использованием трансгенных животных, следует обеспечить прослеживаемость источника таких животных.
- Особое внимание следует уделять выполнению требований нормативных правовых актов Российской Федерации, регулирующих вопросы защиты животных, используемых в научных целях, Виварии для животных, используемых для производства и контроля качества биологических активных субстанций и лекарственных средств, должны быть отделены от зон производства и контроля качества. 24 Для различных видов животных следует определить, контролировать и регистрировать основные показатели. Эти показатели могут включать возраст, вес и состояние здоровья животных.
- Следует предусмотреть обозначения животных, биологических агентов и результатов испытаний для предотвращения риска перепутывания и всех известных опасностей.
Документация
- Может потребоваться включение в документацию на сырье и исходные материалы дополнительных данных об источнике, происхождении, цепи поставок, методах производства и контроля качества для обеспечения необходимого объема контроля, в том числе микробиологического.
- Для некоторых типов продукции может потребоваться специальное описание материалов, входящих в серию, в особенности клеток. Для аутологичных материалов и материалов от специально подобранных доноров продукт следует рассматривать как одну серию.
- При использовании клеток человека или тканей доноров должна быть обеспечена полная прослеживаемость информации, начиная от сырья и исходных материалов, включая все субстанции, вступающие в контакт с клетками иди тканями вплоть до подтверждения приемки продуктов в месте использования, при этом следует соблюдать конфиденциальность всех данных о лицах и здоровье. Документация, отражающая прослеживаемость, должна храниться в течение 30 лет после истечения срока годности лекарственного средства. Особое внимание следует уделять прослеживаемости данных о лекарственных средствах для специальных целей, например, в случае специально подобранного донора. Производство лекарственных средств с использованием компонентов крови в качестве сырья или исходных материалов должно соответствовать установленным требованиям в плане прослеживаемости требований и уведомления о серьезных отрицательных реакциях.
Производство
- Учитывая высокую изменчивость свойств биологических активных субстанций и лекарственных средств, следует обеспечить устойчивость процесса, снижая его вариабельность и повышая воспроизводимость на различных этапах жизненного цикла. При анализе качества продукции следует повторно рассматривать разработку процесса.
- Поскольку условия культивирования, питательные среды и реагенты предназначенные для обеспечения роста клеток и микроорганизмов, являются аксеническими (не содержащими других живых организмов), следует уделить особое внимание мерам по надежному предотвращению или сведению к минимуму нежелательного биозагрязнения и связанных с ним метаболитах и эндотоксинах. В производстве лекарственных средств на основе клеток и тканей, когда серии продукции часто являются малыми, следует установить контроль (требования и инструкции) риска перекрестных загрязнений клеточных препаратов от разных доноров с различным состоянием здоровья.
Сырье и исходные материалы
- Следует четко определять источник, происхождение и пригодность биологического сырья и исходных материалов (например, криопротекторы, питающие клетки, реагенты, питательные среды, буферы, сыворотки, ферменты, цитокины, факторы роста). Если проведение необходимых испытанийзанимает длительное время, допускается начинать обработку исходных материалов до получения результатов испытаний, но следует учесть риск использования дефектных материалов и их влияние на другие серии согласно принципам анализа рисков. В таких случаях выдача разрешения на выпуск серии готовой продукции зависит от результатов этих испытаний. Идентификацию всех исходных материалов следует выполнять исходя из требований к соответствующим стадиям производства. Дополнительные требования для биологических лекарственных средств установлены в части I и приложении 8, а для биологических активных субстанций – части II настоящего стандарта.
- При оценке рисков загрязнения сырья и исходных материалов во время их прохождения по цепи поставки следует уделять особое внимание риску, связанному с губчатой энцефалопатией животных и латентными вирусами. Следует также уделить внимание исходным материалам, вступающим в непосредственный контакт с технологическим оборудованием, и такими веществами как питательные среды, используемые для аттестации асептического наполнения, и смазочные материалы, которые также могут вступать в контакт с продуктом.
- Исходя из того, что существуют риск внесения загрязнений на разных стадиях производства и с соответствующими последствиями для готовой продукции, следует обеспечить защиту продукции, процессов приготовления растворов, буферов и других добавок на основе приложения 1 к настоящему стандарту. Особую роль играет контроль качества сырья и исходных материалов и проведения асептических процессов, особенно для продукции, не подлежащей финишной стерилизации. Если в регистрационном досье или в протоколе клинических исследований установлены допустимый тип и уровень бионагрузки, например, на стадии получения активной субстанции, то стратегия контроля должна предусматривать, за счет чего бионагрузка не выходит за допустимый предел.
- Если требуется стерилизация сырья и исходных материалов, ее следует выполнять термическим методом. При необходимости для инактивации биологических материалов могут использоваться и другие методы, например, радиационная стерилизация и фильтрация.
- Могут потребоваться другие меры, например, использование антибиотиков на ранних стадиях производства, для снижения бионагрузки, связанной с живыми ткаными и клетками. Следует избегать этих мер, но при необходимости их применение должно быть обосновано и они должны быть удалены на стадии, указанной в регистрационном досье или протоколе клинических исследований.
- Получение от донора, закупка и контроль тканей и клеток человека, используемых в качестве сырья или исходных материалов должны выполняться в соответствии с установленными требованиями. Следует обеспечить прослеживаемость тканей и клеток человека, используемых в качестве исходных материалов для биологических лекарственных средств, от донора до серии готового лекарственного средства. Следует предусмотреть организационные меры, регулирующие отношения между производителем и поставщиком тканей и клеток в отношении передачи информации о здоровье до, которая может стать доступной после поставки исходного материала и которая может оказать влияние на качество и безопасность лекарственного средства, получаемого из них.
- Следует обеспечить выполнение требований к прослеживаемости, начиная от поставщика тканей к потребителю и обратно, включая материалы, вступающие в контакт с клетками и тканями;
- Следует заключить техническое соглашение между ответственными сторонами (например, производителями, поставщиками тканей, спонсорами, держателями регистрационного досье), которые определяют задачи каждой стороны, включая ответственное лицо и уполномоченное лицо.
- При использовании в производственных процессах клеток человека или животного в качество следует обеспечить контроль источников, испытаний, транспортирования и хранения, включая контроль в соответствии с требованиями норм к получению от донора, закупке и контролю испытаниям, и законодательства Российской Федерации.
Система посевной культуры и банка клеток
- Для предотвращения нежелательного изменения свойств, в результате многократных пересевов или большого числа генераций, производство биологических лекарственных субстанций и лекарственных средств, получаемых из культур микроорганизмов, культур клеток или размножением в эмбрионах и в животных, должно быть основано на системе главного и рабочих вирусных посевных культур и/или банков клеток.
- Количество генераций (удвоений, пассажей) между посевной культурой или банком клеток и биологической лекарственной субстанцией либо готовым продуктом должно соответствовать требованиям спецификаций в регистрационном досье или протоколе клинических исследований.
- Создание систем посевных культур и банков клеток, включая главные и рабочие генерации посевные культуры, является частью жизненного цикла продукции и должно выполняться в условиях, соответствие которых требованиям ясно подтверждается. К этому относится контролируемая окружающая среда, обеспечивающая безопасность посевных культур и банков клеток работающий с ними персонал.
- После формирования главного и рабочих банков клеток и главного и рабочих банков посевных культур следует выполнять требования к карантину и разрешению банков к использованию. Следует дать адекватное описание и выполнять контроль загрязнений. Пригодность банков к дальнейшему использованию должна быть подтверждена стабильностью характеристик и качеством успешных серий продукции. Доказательство стабильности и воспроизводимости посевных культур или банков следует оформлять документально, организовать хранение документации и анализ тенденций.
- Хранение и использование посевных культур и банков клеток должно быть организовано так, чтобы свести к минимуму риск загрязнений (например, хранить в герметичных контейнерах в парах жидкого азота в герметичных контейнерах) или изменения. При хранении различных посевных культур и/или клеток в одних и тех же зонах или с использованием одного и того же оборудования следует принять меры по предотвращению перепутывания и перекрестного загрязнения с учетом инфекционной природы материалов.
- Емкости для хранения должны быть герметично закрыты и четко маркированы; их необходимо содержать при заданной температуре. Следует вести документальный учет хранящихся емкостей. Температуру хранения следует непрерывно регистрировать, а в установках с жидким азотом контролировать его уровень. Отклонения параметров хранения от установленных пределов и любые предпринятые корректирующие и предупреждающие действия должны быть оформлены документально.
- Рекомендуется разделять запасы на части и хранить их раздельно во избежание полной утраты. Контроль в местах хранения должен обеспечивать выполнение указанных выше требований.
- Условия хранения и обращения с запасами должны определяться согласно тем же требованиям к методам и параметрам. После перемещения контейнеров из хранилища посевной культуры или банка клеток не допускается возвращать их в хранилище повторно.
Принципы работы
- При периодическом внесении изменений следует учитывать их влияние, в том числе всех сторон (например, процесса) на качество, безопасность и эффективность готовой продукции.
- Критические эксплуатационные (технологические) или другие исходные параметры, влияющие на качество продукции, должны быть определены, аттестованы (валидированы), документированы и поддерживаться в соответствии с заданными требованиями.
- Порядок контроля предметов и материалов, поступающих в производственные зоны, должен основываться на принципах анализа рисков для качества. Для асептических процессов термостойкие предметы и материалы, поступающие в чистую или чистую и изолированную зону должны, по возможности, проходить обработку в проходном автоклаве или сухожаровом шкафе (туннеле). Нетермостойкие предметы и материалы должны поступать через воздушные шлюзы с блокировкой дверей* и проходить эффективную обработку (дезинфекцию) поверхностей. Допускается стерилизация предметов и материалов в другом месте при условии, что число слоев обертки (упаковки) соответствует числу стадий при поступлении в чистую зону, причем они перемещаются в чистую зону через воздушные шлюзы с обработкой (дезинфекцией) поверхностей.
- Следует подтвердить ростовые свойства питательных сред в соответствии с показателями назначения. Питательные среды должны, по возможности, проходить стерилизацию на месте. Следует предусматривать, если это возможно, стерилизующие фильтры в системах подачи газов, сред, кислот или щелочей, пеногасителей к ферментерам.
- Добавление веществ или культур в ферментеры и другие сосуды, а также отбор проб из них следует проводить в тщательно контролируемых условиях для предотвращения загрязнений. При внесении добавок или отборе проб следует контролировать правильность подсоединения сосудов.
- При необходимости может выполняться непрерывный контроль некоторых производственных процессов (например, ферментации) с внесением результатов контроля в протоколы серий. При непрерывном культивировании следует выполнять специальные требования к контролю качества исходя из вида процесса.
- Процессы центрифугирования и смешивания продуктов могут приводить к образованию аэрозолей, поэтому для сведения к минимуму перекрестных загрязнений их следует проводить в изолированных зонах.
- При случайной утечке, в особенности живых организмов, следует принять неотложные меры безопасности. Для каждого вида или группы организмов следует предусмотреть аттестованные методы дезинфекции (стерилизации). При использовании различных штаммов бактерий одного вида или очень похожих вирусов эта процедура может быть аттестована только для одного штаммапредставителя, если у них нет существенных различий в устойчивости к соответствующему методу дезинфекции (стерилизации).
- При явном наличии загрязнений, например, при проливах или следах аэрозолей или наличии потенциально опасных организмов, следует дезинфицировать материалы (в т. ч. для ведения документации), которые используются в производстве и при контроле, или применять другие носители информации.
- При инактивации или удалении вирусов в ходе производства следует избегать риска повторного загрязнения обработанной продукции со стороны необработанной продукции.
- Для продуктов, инактивируемых при помощи добавления реагентов (например, микроорганизмы в процессе производства вакцин), процесс должен гарантировать полную инактивацию живых микроорганизмов. В дополнение к тщательному смешиванию культуры и инактивирующего агента следует обращать внимание на контакт с живой культурой всех поверхностей, вступающие в контакт с продуктом, и, при необходимости, на передачу в другой сосуд.
- При применении хроматографических методов используются разные виды оборудования. При разработке порядка контроля сорбентов, корпусов колонок и другого оборудования в случае производства циклами или многономенклатурного производства следует применять принципы анализа рисков для качества. Не рекомендуется использование одних и тех же матриц на разных технологических стадиях. Следует установить критерии приемлемости, условия работы, методы восстановления, срок службы и методы стерилизации или дезинфекции колонок.
- Работа с облученными оборудованием и материалами рассмотрена в приложении 12 к настоящему стандарту.
- Следует предусмотреть меры, обеспечивающие целостность и герметичность контейнеров после их наполнения, если готовая или промежуточная продукция представляет собой особый риск, и утвердить инструкции по действиям в случае утечек или проливов. Для операций наполнения и упаковки следует иметь инструкции по соблюдению требований к нахождению продукции в заданных пределах параметров (например, к времени и (или) температуре).
- Работа с флаконами, содержащими биологические агенты, должна проводиться таким образом, чтобы избежать загрязнения другой продукции или проникания живых агентов в производственную или внешнюю среду. Следует учитывать жизнеспособность таких организмов и их биологическую классификацию, исходя из фактора риска.
- Следует выполнять меры предосторожности при изготовлении, печати, хранении и использовании этикеток, включая специальные тексты, помещаемые на первичной и наружной упаковке продуктов, специфических для пациента.
- При использовании сверхнизких температур хранения следует подтвердить устойчивость маркировки к таким температурам
- Если данные о состоянии здоровья донора (человека или животного), имеющие значение для качества продукции, становится доступной после закупки, это должно учитываться в инструкции по отзыву продукции.
Контроль качества
- Внутрипроизводственный контроль играет более важную роль в обеспечении неизменности качества биологических активных субстанций и лекарственных средств, чем для обычной продукции. . Внутрипроизводственный контроль должен осуществляться на соответствующих стадиях производства с целью контроля условий, являющихся важными для качества готовой продукции.
- Если промежуточная продукция может храниться в течение длительного времени (дни, недели или более), то следует рассмотреть возможность включения в текущую программу испытания стабильности серий готовой продукции, которые произведены из промежуточной продукции с максимальным периодом хранения в процессе производства.
- Некоторые типы клеток (например, аутологичных клеток) могут быть доступны в ограниченных количествах и для них может быть изменен порядок испытаний и хранения контрольных образцов документальным оформлением, если это допускается регистрационным свидетельством.
- Для продукции на основе клеток испытания на стерильность следует проводить на культурах клеток или банках клеток, свободных от антибиотиков, чтобы получить доказательства отсутствия загрязнения бактериями и грибами, и иметь возможность обнаружения организмов, требующих специальных условий культивирования (если применимо).
- Для биологических лекарственных средств с малым сроком годности (определяемым в настоящем Приложением как срок до 14 дней) и необходимости выпуска серии до завершения всех видов контроля качества (например, испытаний на стерильность), следует ввести специальный порядок контроля.
Часть В. Специальное руководство по отдельным вилам продукции
В1. Продукция животного происхождения
Настоящее руководство распространяется на материалы животного происхождения, в том числе материалы, полученные из таких предприятий, как скотобойни. Поскольку цепи поставок могут быть обширными и сложными, следует организовать контроль на основе принципов анализа рисков. При этом необходимо учитывать требования Государственной фармакопеи Российской Федерации, включая проведение соответствующих испытаний на определенных стадиях. Следует вести документацию, позволяющую проследить цепь поставок, с достаточно подробным описанием роли каждого участника цепи поставок и схему поставок.- Следует иметь программы контроля опасных для человека болезней животных (ветеринарного освидетельствования). При оценке факторов риска и сведении их к минимуму учитываются данные от заслуживающих доверие источников о распространении заболевания на территории страны. Такая оценка проводится организациями, к которым относится Всемирная организация по охране здоровья животных (Международное Эпизоотическое Бюро – МЭБ). Это должно сопровождаться данными о контроле здоровья и программах контроля на национальном и местном уровнях, включая контроль источников (например, ферм или загонов для скота), из которых получены животные, и контроль при транспортировании животных на скотобойню.
- Скотобойни должны соответствовать требованиям, установленным нормативными правовыми актами Российской Федерации, при использовании их в качестве поставщиков тканей животных. Следует учитывать сведения от уполномоченного федерального органа исполнительной власти, подтверждающие соблюдение требований безопасности и качества кормов, установленные нормативными правовыми актами Российской Федерации и (или) других стран, из которых сырье импортируется в Российскую Федерацию.
- Меры по контролю сырья и исходных материалов на таких предприятиях, как скотобойни, должны включать элементы системы качества для обеспечения требуемой подготовки персонала, прослеживаемости материалов, контроля и стабильности. Эти меры могут быть заимствованы из источников, не относящихся к данному стандарту, но они должны обеспечить эквивалентный уровень контроля.
- Следует предусмотреть меры по контролю сырья или исходных материалов, обеспечивающие предотвращение вмешательств, влияющих на качество материалов, или, по меньшей мере, содержать информацию о таких мерах при движении материалов по цепи производства и поставки. К этому относится движение материалов от мест первичного сбора, проведения частичной и полной очистки до мест хранения, накопления, размещения и нахождения у посредников. Следует подробно оформлять проведение этого процесса, обеспечивая прослеживаемость, и регистрировать любые нарушения с их расследованием и принятием мер.
- Следует регулярно проводить оценку поставщиков сырья и исходных материалов с подтверждением выполнения требований к их контролю на разных стадиях производства. Следует детально анализировать отклонения с учетом из значимости с полным документальным оформлением. Должна быть также предусмотрена система внесения корректирующих и предупреждающих действий.
В2. Препараты от аллергии
Исходные материалы могут быть получены методом извлечения из естественных источников или с использованием технологии рекомбинантных ДНК.- Следует дать достаточно подробное описание источника для обеспечения надежности поставок, включая, например, общепринятое и научное название, происхождение, природу, допустимые пределы загрязнений и метод получения. Материалы животного происхождения должны быть получены от здоровых животных. Следует организовать биологический контроль колоний (например, клещей, животных), используемых для экстракции аллергенов. Препараты от аллергии должны храниться в соответствующих условиях, обеспечивающих их качество.
- Следует дать подробное описание и провести аттестацию (валидацию) стадий технологического процесса, включая предварительную обработку, экстракцию, фильтрацию, диализ, концентрирование или лиофилизацию.
- Следует давать описание процессов модификации, используемых в производстве модифицированных экстрактов аллергенов (например, аллергоидов, конъюгатов). Следует давать описание и контролировать промежуточные продукты, получаемые в технологическом процессе.
- Смеси экстрактов аллергенов должны быть приготовлены из отдельных экстрактов исходных материалов, полученных из одного источника. Каждый отдельный экстракт должен рассматриваться как отдельная активная субстанция.
В3. Продукция на основе иммунных сывороток животных
- Следует уделять особое внимание контролю антигенов биологического происхождения для гарантии их качества, постоянства и отсутствия посторонних включений. Подготовка материалов для иммунизации животных (например, антигенов, гаптен-носителей, адъювантов, стабилизаторов) и их до иммунизации должно выполняться в соответствии с документаций.
- Порядок иммунизации, исследований и отбора крови должен соответствовать регистрационному досье.
- Условия при приготовлении субфрагментов антител (например, участки связывания антигена Fab или F(ab’)2) и любые дальнейшие модификации должны соответствовать аттестованным (валидированным) и утвержденным параметрам. Если такие энзимы состоят из нескольких компонентов, то следует обеспечить их стабильность.
В4. Вакцины
- При использовании яиц следует обеспечить здоровье всех стай, которые служат для их получения (стай, свободных от специфических патогенов, и здоровых стай).
- Следует проводить аттестацию (валидацию) целостности контейнеров, используемых для хранения промежуточных продуктов, и времени их хранения.
- Не допускается открывание сосудов, содержащих инактивированные продукты, или отбор проб из них в зонах, где находятся живые биологические агенты.
- Последовательность прибавления активных компонентов, адъювантов и вспомогательных веществ в процессе производства промежуточного или готового продукта должна соответствовать технологическим инструкциям.
- Следует обеспечить необходимые меры изоляции при использовании для производства или испытаний микроорганизмов, которым присвоен высший уровень биологической опасности (например, пандемические штаммы). Разрешения на проведение мероприятий должно быть получено от компетентного органа. Разрешительная документация должна быть в наличии на случай проверки.
В5. Рекомбинантные продукты
- Следует выполнять требования, предъявляемые к условиям для роста клеток, синтезу белков и процессам очистки в пределах заданных параметров для обеспечения постоянства свойств продукции и соответствия допустимым пределам для примесей, причем эти требования должны учитывать возможности процесса находиться в указанных пределах. Для типа клеток, используемых в производстве, могут потребоваться более жесткие меры для гарантии отсутствия вирусов.
- Процессы очистки от нежелательных белков клеток хозяина, в частности от белков, нуклеиновых кислот, углеводов, вирусов и других примесей, следует проводиться в рамках заданных и подтвержденных пределов.
В6. Моноклональные антитела
- Моноклональные антитела могут быть произведены из гибридов мышей или человека с помощью технологий рекомбинантных ДНК. Для обеспечения безопасности и качества продукции следует контролировать исходные клетки (в том числе, питающие клетки, если применяются) и исходные материалы, используемые для создания гибридомы и (или) линии клеток. Необходимо удостовериться, что данные операции проводятся в утвержденных пределах. Особое внимание должно уделяться доказательству отсутствия вирусов. Для доказательства пригодности лекарственных средств, произведенных на одной и той же технологической основе, возможно использование данных, полученных при испытании одного из них.
- Следует контролировать соответствие заданным значениям параметров на промежуточной и завершающей стадиях технологического процесса.
- Условия производства при приготовлении суб-фрагментов антител (например, Fab, F(ab’)2, scFv) и любых других модификаций (например, введения радиоактивных меток, конъюгации, химического связывания) должны соответствовать заданным параметрам.
В7. Лекарственные препараты трансгенных животных
Обеспечение постоянства исходного материала, полученного из трансгенного источника, является более сложным, чем при использовании стандартных нетрансгенных биотехнологических источников. Поэтому к подтверждению неизменности свойств продукции от серии к серии предъявляются повышенные требования.- Для производства биологических лекарственных средств могут использоваться различные виды животных, в том числе могут проводиться получение и очистка биологических жидкостей (например, молока). Животные должны иметь четкую и уникальную маркировку, и должны быть предусмотрены дублирующие меры на случай утраты первичного идентифицирующего маркера.
- Условия содержания и ухода за животными должны обеспечивать наименьший возможный контакт животных с патогенными организмами и зоонозами. Следует предусмотреть необходимые меры по защите окружающей среды. Должна быть разработана программа наблюдения за здоровьем животных с соответствующим внесением записей в документацию. Также должны быть расследованы любые отклонения и определено их влияние на возможность дальнейшего использования животного и ранее полученных серий продукции. Необходимо удостовериться, что любые лекарственные средства, применявшиеся для лечения животных, не приведут к загрязнению выпускаемого лекарственного средства.
- Родословная от животного-основателя до животных, использующихся для производства, должна быть оформлена документально. Запрещается смешивание материалов, полученных от разных трансгенных линий животных, поскольку они происходят от разных животных-основателей.
- Условия, при которых происходит получение материалов, должны соответствовать регистрационному досье и протокола клинических исследований. График получения материала и условия, при которых животные могут быть исключены из процесса производства продукции, должны соответствовать утвержденной документации и заданным параметрам.
В8. Продукция из трансгенных растений
Обеспечение постоянства исходного материала, полученного из трансгенного источника, является более сложным, чем при использовании стандартных нетрансгенных биотехнологических источников. Поэтому подтверждению неизменности свойств продукции от серии к серии предъявляются повышенные требования.- Для предотвращения загрязнения главных и рабочих трансгенных банков посторонними материалами растительного происхождения и соответствующими посторонними агентами могут понадобиться дополнительные меры по сравнению с разделом А. Контроль стабильности гена должен проводиться на протяжении определенного количества генераций.
- Для обеспечения постоянства сбора урожая от разных культур растений растения должны иметь четкую и уникальную маркировку. Следует контролировать основные показатели, включая состояние здоровья растений данной культуры, с определенной периодичностью на протяжении периода выращивания для обеспечения их постоянства для разных посевов.
- Следует предусмотреть меры предосторожности для защиты культур. По возможности нужно свести к минимуму микробиологическое загрязнение и перекрестные загрязнения растениями другого вида. Следует принять меры по предотвращению загрязнения продукции такими веществами, как пестициды и удобрения. Должна быть разработана программа контроля с внесением записей в документацию, также должны быть расследованы любые отклонения и определено их влияние на возможность дальнейшего использования культуры в производстве.
- Следует четко определить условия, при которых растения могут быть исключены из производственного процесса. Необходимо установить пределы пригодности материалов (например, основных белков), которые могут помешать процедуре очистки продукции. Следует подтвердить соответствие результатов заданным пределам.
- Следует регистрировать параметры окружающей среды (температура, выпадение осадков), которые могут оказать влияние на показатели качества лекарственного препарата и на выход рекомбинантного белка в производстве, начиная от времени посева, на протяжении культивирования и до момента сбора и промежуточного хранения собранных материалов. При оформлении указанной документации должны соблюдаться требования нормативных правовых актах Российской Федерации, регулирующих вопросы выращивания и сбора растений.
Термины и определения к приложению 2
Ниже приводятся только термины, используемые в приложении 2 и требующие пояснения. Для терминов, определения которым даны в нормативных правовых документах или в других источниках даны ссылки. В дополнение к этим терминам, используются общие термины по GMP данного стандарта, если не указано иное. Активная субстанция (active substance): См. раздел 20 части II. Стимулятор (adjuvant): химическое и биологическое вещество, усиливающее иммунную реакцию на антиген. Аллергоид (allergoid): химически модифицированный аллерген, снижающий реактивность на общий иммуноглобулин Е (lgE). Антиген (antigen): вещество (например, токсины, чужеродные белки, бактерии, клетки тканей) способное вызвать реакцию иммунной системы. Антитело (antibody): белковое соединение, вырабатываемое В-лимфоцитами, которые связываются с определенными антигенами. Антитела могут быть разделены на два основных типа в зависимости от существенных различий в методе производства. Моноклональные антитела (monoclonal antibodies – MAb): однородная популяция антител, полученная из одного клона лимфоцитов или по рекомбинантной технологии и которая связывается с одним эпитопом (антигенной детерминантой). Поликлональные антитела (polyclonal antibodies): антитела, получаемые из разных клонов лимфоцитов, вырабатываемые в человеке и животных в ответ на эпитопы на большинстве чужеродных молекул. Зона (area): Определенный комплекс помещений в здании, относящихся к производству одного продукта или разных продуктов, который обслуживается одним кондиционером. Бионагрузка (bioburden): количество и тип (например, недопустимые или допустимые) микроорганизмов, находящихся в сырье, среде, биологическом веществе, промежуточной или готовой продукции. Рассматривается как загрязнение, если их количество и/или вид выходит за установленные пределы. Биологическое лекарственное средство (biological medicinal product): лекарственное средство с активной субстанцией биологического происхождения. Уровень биобезопасности* (biosafety level – BSL): Условия изоляции, требуемые для безопасного обращения с организмами, имеющими степени опасности от BSL1 (низкий риск, заболевание человека маловероятно) до BSL4 (высший риск, вызывает серьезную болезнь, распространение которой вероятно и против которой отсутствуют профилактические меры и лечение). Производство циклами (campaign manufacture): Производство серий одного и того же продукта в последовательном порядке в течение заданного периода времени в четком соответствии с принятыми мерами контроля до перехода к производству другого продукта. Разные продукты не должны производиться одновременно, но могут производиться на одном оборудовании Банк клеток (cell bank): совокупность контейнеров, содержание которых однородно по составу и которые хранятся при определенных условиях. Каждый контейнер содержит аликвоту (часть) одной культуры клеток. Клеточный запас (cell stock): первичные клетки, размножившиеся до заданного количества клеток, подлежащие разделению на аликвоты (части) и использования в качестве исходного материала для производства ограниченного количества серий лекарственных препаратов на основе клеток. Закрытая система (closed system): Система, в которой субстанция лекарственного средства и продукт не оказывается открытым в непосредственно окружающую ее среду помещения в ходе производства. Использование условий изоляции (contained use): деятельность, при которой генетически модифицированные организмы получают, культивируют, хранят, транспортируют, разрушают, уничтожают или используют каким-либо способом с применением мер по изоляции для ограничения распространения этих организмов и обеспечения безопасности населения и охраны окружающей среды. Преднамеренный выброс (deliberate release): преднамеренный выпуск в окружающую среду генетически модифицированных организмов, для которых не используются специальные меры изоляции для ограничения распространения этих организмов и обеспечения безопасности населения и окружающей среды. Вспомогательное вещество (excipient): вещества неорганического или органического происхождения, используемые в процессе производства лекарственных средств, не являющиеся активными субстанциями. Вне живого организма, ex-vivo (ex-vivo): процесс, происходящий на тканях или клетках вне живого организма и возвратом их в живое тело. Питающие клетки (feeder cells): клетки, используемые при совместном культивировании для поддержания плюрипотентных стволовых клеток. Для культуры стволовых клеток эмбриона человека типичные питающие слои включают фибробласты эмбрионов мышей (MEFs) или фибробласты эмбрионов человека, которые были обработаны для предотвращения деления. Ген (gene): последовательность ДНК, содержащая коды для одного или более белков. Генетически модифицированный организм, ГМО (genetically modified organism – GMO): любой организм, за исключением организма человека, в котором генетический материал был изменен таким образом, что достичь такого изменения в результате естественного скрещивания и (или) естественной рекомбинации невозможно. Гаптен, неполноценный антиген (hapten): молекула с низкой молекулярной массой, которая сама не являющаяся антигеном, если она не конъюгирована (не сопряжена) с «молекулойносителем». Гибридома (hybridoma): «бессмертная» (способная делиться большое количество раз) линия клеток, которые выделяют желаемые (моноклональные) антитела, и обычно получаемая искусственным слиянием В-лимфоцитове с клетками опухоли. Промежуточный продукт (intermediate product): Частично обработанный материал, который должен пройти дальнейшие стадии производства, прежде чем он станет нерасфасованным готовым продуктом. Внутри живого организма, in-vivo (in-vivo): процессы, проходящие внутри живого организма. Ретроспективный анализ (look-back): документально оформленные действия по прослеживанию субстанций биологических лекарственных средств и продукции, качество которых может быть ухудшено при использовании или введении материалов животного или человеческого происхождения, если контроль качества при выпуске таких материалов выявил загрязнения или если очевидно несоответствие у животных или человека, от которых эти материалы получены. * Перевод определения термина «уровень безопасности» дан по оригиналу правил GMP EC. Действующая в России классификация микроорганизмов по степени опасности зеркально противоположна применяемой в мире и в Европе классификации, т. е. в России микроорганизмы с высшим риском относятся к группе 1, а с низшим – к группе 4, что вызывает системную путаницу (прим. разработчика стандарта). Главный банк клеток (master cell bank – MCB): Аликвота (часть) одной культуры клеток, приготовленной, как правило, из выбранного клеточного клона при определенных условиях, распределенная по разным контейнерам и хранимая при определенных условиях. Используется для получения всех рабочих банков клеток. Главный вирусный посев (master virus seed – MVS): указанный выше банк, но относящийся к вирусам. Главный трансгенный банк (master transgenic bank) – указанный выше банк, но относящийся к трансгенным растениям или животным. Чистая культура (monosepsis (axenic)): Культура, содержащая единственный микроорганизм, не загрязненная любыми другими организмами. Многономенклатурное производство (multi-product facility): производство, которые выпускает последовательно или циклами разные биологические субстанции лекарственных средств и продукты и внутри которого комплекс оборудования может быть или может не быть специализирован для производства определенной субстанции или продукта. Плазмида (plasmid): часть ДНК, обычно существующая в бактериальной клетке в виде кольцевой структуры, отделенная от клеточных хромосом. Может быть модифицирована с помощью методов молекулярной биологии, выделена из бактериальной клетки и использована для переноса ее ДНК в другую клетку. Сырье(raw materials):материалы, но проходившие переработку. Ответственное лицо (responsible person): специально назначенное лицо в организации по производству биологических активных субстанций и лекарственных препаратов, которое несет ответственность за:- обеспечение того, что биологический материал (в том числе клетки и (или) ткани человека) получен, проверен, использован в процессе производства лекарственного средства включая контроль качества готового продукта, а также хранился и был отпущен в соответствии с законодательством Российской Федерации;
- предоставление уполномоченным федеральным органам исполнительной власти необходимой информации относительно предписаний, разрешений, аккредитации или лицензирования;
- выполнение в организации по производству биологических (в том числе иммунобиологических) фармацевтических субстанций и лекарственных препаратов, всех требований законодательства Российской Федерации.
Введение
Эту форму следует использовать совместно с Делегированным регламентом ЕС No 1569/2017 по Правилам производства (GMP) лекарственных средств для исследований (IMP), предназначенных для человека, и организацией инспектирования, которое имеет законную силу в соответствии с первым подпунктом статьи 63(1) Регламента ЕС No 536/2014 и подробным Руководством Комиссии по GMP для IMPs, предназначенных для применения человеком в соответствии со вторым подпунктом статьи 63(1) Регламента ЕС No 536/2014. Лекарственные средства для исследований могут не использоваться в клинических испытаниях в стране-члене ЕС до завершения двухстадийной процедуры в соответствии с «Руководством по ответственности спонсора в отношении обращения и отгрузки лекарственных средств для исследований, предназначенных для человека, в соответствии с Правилами проведения клинических испытаний (GCP) и Правилами производства лекарственных средств (GMP)». Первым этапом является сертификация каждой серии Уполномоченным лицом производителя или импортера в соответствии со статьей 62(1) Регламента ЕС No 536/2014 для подтверждения соответствия требованиям статей 63(1) и 63(3) Регламента ЕС No 536/2014 и статьи 12 Делегированного регламента комиссии ЕС No 1569/2017 с документальным оформлением. Для обеспечения свободного перемещения лекарственных средств для исследований между странами-членами сертификация серии за подписью Уполномоченного лица должна соответствовать статье 62(1) Регламента 536/2014. Содержание этих сертификатов должно соответствовать приводимой ниже форме с целью гармонизации процесса сертификации при выпуске серии. Эта форма может также использоваться для сертификации серий, предназначенных для использования в стране-члене по месту нахождения производителя или импортера. [Головка бланка письма производителя]Форма сертификата серии лекарственных средств для исследований в соответствии со статьей Article 62(1) Регламента ЕС No 536/2014 и статьей 4 Делегированного регламента 1569/2017
- Наименование продукта(ов)/идентификационного обозначение продукта(ов) согласно заявлению на клинические испытания, если требуется.
- Номер(а) в базе данных препаратов для клинических испытаний в ЕС (EudraCT) и протокола спонсора, если требуется
- Эффективность
- Дозированная форма (фармацевтическая форма)
- Размер упаковки (содержание контейнера) и вид (например, флаконы, бутылки, блистеры)
- Номер серии/партии
- Срок годности/повторного контроля/использования
- Наименование и адрес производителя, где находится Уполномоченное ли- цо, выдавшее сертификат
- Номер лицензии на производство для производственной площадки по п. 8
- Комментарии/примечания
- Любая дополнительная информация, которую сочло нужным внести Уполномоченное лицо
- Заключение о сертификации
- «Настоящим я подтверждаю, что данная серия соответствует требованиям статьи 62(1) Регламента ЕС No 536/2014 и статьи 4 Делегированного регламента 1569/2017»
- Имя Уполномоченного лица, выдавшего сертификат
- Подпись
- Дата подписи
Введение
Данное руководство основано на пятом параграфе статьи 47 Директивы 2001/83/ЕС1. В соответствии со вторым параграфом статьи 46(f) Директивы 2001/83/ЕС держатель лицензии на производство должен обеспечить пригодность вспомогательных веществ для производства лекарственных средств, установив объем внедрения правил GMP. Требования GMP для вспомогательных веществ, предназначенных для производства лекарственных средств для человека, должны основываться на формализованной оценке рисков в соответствии с данным руководством. При оценке рисков следует учитывать требования других относящихся к данному предмету систем качества, а также происхождение и назначение вспомогательных веществ и данные о дефектах качества в прошлом. Держатель лицензии на производство должен обеспечить выполнение соответствующих требований GMP. Держатель лицензии на производство должен включить процесс оценки рисков для вспомогательных веществ в фармацевтическую систему качества. Держатель лицензии на производство процесс должен иметь документацию по оценке рисков в соответствии с применяемыми требованиями GMP, доступную для рассмотрения инспектором по GMP. Следует извещать производителя вспомогательных веществ о результатах оценки рисков для обеспечения постоянного улучшения. Оценка рисков в соответствии с данным руководством должна быть выполнена для вспомогательных веществ для допущенных к применению лекарственных средств для человека не позднее 21 марта 2016 г.Глава 1. Область применения
1. 1 Данное руководство применяется при оценке рисков при внедрении правил GMP для вспомогательных веществ, используемых при производстве лекарственных средств для человека. В соответствии со статьей 1(3b) Директивы 2001/83/ЕС под вспомогательным веществом понимается любое вещество, входящее в состав лекарственного средства, не относящееся к активным субстанциям или упаковочным материалам. 1.2 Данное руководство не распространяется на вещества, добавляемые для обеспечения стабильности активных субстанций, которые не могут существовать самостоятельно.Глава 2. Требования GMP, относящиеся к виду и назначению вспомогательно- го вещества
2.1 Методы анализа рисков, которые можно применять для различных сторон фармацевтического качества, включая вспомогательные вещества, приведены в части III на- стоящего стандарта (Руководство Eudralex, том 4 – правила производства лекарственных средств для человека и животных – GMP EC, часть III), руководстве ICH Q9 по анализу рисков. 2.2 Эти принципы анализа рисков следует использовать при оценке рисков для качества, безопасности и функции каждого вспомогательного вещества и их классификации, например малый риск, средний риск и высокий риск. С этой целью рекомендуется применять методы согласно части III данного стандарта (руководство ICH Q9), например анализ рисков в критических контрольных точках (HACCP). 2.3 Для каждого вспомогательного вещества, получаемого от каждого производите- ля, держатель лицензии на производство должен установить риск, представляющий угрозу для безопасности, качества и функции каждого вспомогательного вещества из этого источника (животного, минерального, растительного, полученного методом синтеза и т. д.) на всем протяжении для введения в готовую дозированную фармацевтическую форму. При этом следует учитывать (перечень не исчерпывающий) следующее:- трансмиссивную губчатую энцефалопатию;
- возможные вирусные загрязнения;
- возможные загрязнения микроорганизмами, эндотоксинами (пирогенами);
- возможность внесения загрязнений от исходных материалов, например, афлатоксинов или пестицидов или полученных в ходе процессами переносимых далее загрязнений, например, остатки растворителей или катализаторы;
- обеспечение стерильности вспомогательных веществ, заявленных стерильными;
- возможности переноса любых загрязнений от других процессов при отсутствии специализированного (выделенного) оборудования и/или помещений;
- контроль окружающей среды и условия хранения и транспортирования, включая холодовые цепи, если требуется;
- сложность цепи поставки;
- стабильность вспомогательного вещества;
- свидетельство о целостности упаковки.
- фармацевтическую форму и назначение лекарственного средства, содержащего данное вспомогательное вещество;
- функцию вспомогательного вещества в производстве, например, смазка в производстве таблеток или консервант жидких форм и т. д.;
- долю вспомогательного вещества в составе лекарственного средства;
- дневной прием вспомогательного вещества пациентом;
- любые известные дефекты качества или фальсификацию, как в глобальном, так в локальном масштабе компании, относящиеся к данному вспомогательному веществу;
- имеет ли вспомогательное вещество сложный состав;
- известное или возможное влияние на критические свойства для качества лекарственного средства;
- другие обнаруженные факторы или факторы, которые могут относиться к обеспечению безопасности.
- разработку и применение эффективной фармацевтической системы качества;
- наличие компетентного или квалифицированного персонала;
- описание выполняемых функций руководящим персоналом, ответственным за производство и контроль качества;
- программы обучения для всего персонала, связанного с производством и качеством;
- программы обучения, относящиеся к здоровью, гигиене и одежде, если необходимо для выполняемых операций;
- обеспечение помещениями и оборудованием для выполнения операций и их техническое обслуживание;
- документально оформленную систему(ы), охватывающую все процессы и требования для различных операций по производству и контролю качества;
- порядок кодирования и идентификации исходных и промежуточных материалов и вспомогательных веществ, позволяющий обеспечить полное прослеживание;
- программу аттестации поставщиков;
- систему контроля качества вспомогательных веществ и лицо, независимое от производства, которые выпускает серию продукции;
- хранение протоколов для всех исходных материалов и вспомогательных веществ и хранение образцов (проб) вспомогательных веществ в течение времени, определенного частью II данного стандарта;
- систему, обеспечивающую выполнение любых работ по контракту в соответствии с письменным контрактом;
- поддержание эффективной системы рассмотрения рекламаций и отзывов вспомогательных веществ;
- систему внесения изменений и контроля отклонений;
- порядок самоинспекций;
- контроль окружающей среды и условия хранения.
Глава 3. Определение степени рисков у производителя вспомогательных веществ
3.1 После определения требований GMP, которые следует выполнять, нужно про- вести анализ фактического выполнения этих требований производителем вспомогательных веществ. 3.2 Данные для такого анализа следует получить путем проведения аудита или из информации от производителя вспомогательных веществ. 3.3 Следует учесть проведение аттестации системы качества и/или оценить выполнение требований GMP и стандартов производителем вспомогательных веществ и на- сколько такая аттестация соответствует требованиям. 3.4 Следует документально оформить любые несоответствия требованиям GMP фактического состояния дел у производителя вспомогательных веществ. Более того, держателю лицензии на производство следует выполнить дальнейшую оценку рисков, чтобы оценить степень риска, например, низкий, средний или высокий для данного производителя вспомогательных веществ. Для этой цели следует использовать руководство ICH Q9 (часть III данного стандарта). 3.5 Держатель лицензии на производство должен иметь различные способы от принятия путем контроля до непринятия для различных степеней риска на основе этих критериев, например, путем проведения аудита, документации по анализу и контролю.Глава 4. Подтверждения заявления о соответствии требованиям GMP
4.1. После определения применимых требований GMP к производству вспомогательных веществ и степеней риска следует выполнить дальнейшее рассмотрение рисков, например, учитывая:- количество дефектов в сериях полученных вспомогательных веществ;
- вид/тяжесть таких дефектов;
- анализ результатов контроля и тенденций изменения качества вспомогательных веществ;
- недостатки в системе качества и/или проведенной аттестации на соответствие GMP производителем вспомогательных веществ;
- анализ тенденций изменения показателей качества лекарственного средства; это может зависеть от природы и назначения вспомогательного вещества;
- оценку изменений в организации работы, методах и технических процессах у производителя вспомогательных веществ;
- результаты аудитов и повторных аудитов производителя вспомогательных веществ;
- вопросники.
1. Введение
При производстве лекарственных средств возможны случайные перекрестные загрязнения в результате неконтролируемого выделения пыли, газов, паров, аэрозолей, генных материалов или организмов из активных субстанций, других исходных мате- риалов и других продуктов, которые производятся одновременно, а также от остатков на оборудовании и одежды операторов. Исходя из понимания риска, ранее требовалось выпускать некоторые виды лекарственных средств на специализированном или отдельном изолированном оборудовании (помещении), включая «некоторые антибиотики, некоторые гормоны, некоторые цитотоксины и некоторые высокоактивные лекарственные средства». Как ранее, так и в настоящее время отсутствует официальное руководство, позволяющее производителям проводить различие между отдельными продуктами внутри заданных классов. Пересмотр глав 3 и 5 правил GMP выполнен с целью применения научного подхода и подхода, основанного на анализе рисков, и введено понятие «токсикологическая оценка» при определении пороговых величин для идентификации риска. Очистка является мерой по снижению риска и в фармацевтической промышленности широко используются пределы переноса при исследованиях во время аттестации процессов очистки. Для установления этих пределов используются разные подходы, которые не всегда учитывают доступные фармакологические и токсикологические данные. Следовательно, нужен более научный подход для отдельных случаем при идентификации риска и определения мер по снижению риска для всех классов фармацевтических субстанций. Данное руководство содержит рекомендации по рассмотрению и оценке фармакологических и токсикологических данных для отдельных активных субстанций и таким образом позволяет определить пороговые уровни в соответствии с правилами GMP. Эти методы могут использоваться для идентификации рисков и обоснования пределов переноса при валидации процессов очистки. В то время, как активные фармацевтические субстанции (АФС) не рассматриваются в главах 3 и 5 правил GMP, для определения пороговых величин при идентификации рисков могут быть использованы общие принципы данного руководства, где требуется. Допускается отклонение от основного подхода, приведенного в настоящем руководстве, для определения таких пределов безопасности, если они адекватно обоснованы.2. Область применения
Данное руководство предназначено для обеспечения безопасности людей и целевых животных, которые подвергаются воздействию остатков активных субстанций в лекарственных средствах, а также возможных потребителей остатков активных субстанций, находящихся в продуктах питания животного происхождения в результате лечения животных, используемых для питания, ветеринарными препаратами, в которых находятся остатки активных субстанций. Таким образом, данный документ рекомендует подходы к получению научно обоснованных пределов для отдельных активных субстанций, которые используются для идентификации риска. Руководство показывает, как данные, на основе которых получен предел, могут использоваться для формирования ясного и гармонизированного подхода для фармацевтической промышленности.3. Нормативные ссылки
Данное руководство следует использовать совместно со следующими документами: EudraLex – Volume 4 Good manufacturing practice (GMP) Guidelines, Chapter 3 and 5 (Правила GMP EC – Правила производства и контроля качества лекарственных средств. Главы 3 и 5) Note for Guidance on Impurities: Residual Solvents (CPMP/ICH/283/95 in conjunction with CPMP/ICH/1507/02, CPMP/ICH/1940/00 corr, CPMP/QWP/450/03, EMEA/CVMP/511/03 and CPMP/QWP/8567/99). VICH GL18(R): Impurities: Residual solvents in new veterinary medicinal products, active substances and excipients (EMA/CVMP/VICH/502/99-Rev.1). Guideline on the Limits of Genotoxic Impurities (EMEA/CHMP/QWP/251344/2006 and CPMP/SWP/5199/02).4. Определение пределов воздействия на здоровье
4.1 Вычисление допустимого дневного воздействия (PDE)
Предлагаемый в данном документе метод по определению пределов воздействия на здоровье для остатков активных субстанций основан на методе установления так называемого допустимого дневного воздействия (PDE) согласно приложению 3 ICH Q3C (R4) «Руководство по остаткам растворителей» («Impurities: guideline for residual solvents») и приложения 3 руководства VICH GL 18 по «остаткам растворителей в новых ветеринарных препаратах, активных субстанциях и вспомогательных веществах (пересмотр)» («residual solvents in new veterinary medicinal products, active substances and excipients (Revision)»). Величина PDE представляет специфичную для субстанции дозу, которая скорее всего не вызовет отрицательный эффект, если индивидуум подвергается воздействию этой или меньшей дозы каждый день в течение жизни. Определение величины PDE включает в себя:- установление опасности на основе всех имеющихся данных;
- установление «критических эффектов»;
- определение уровня, при котором не наблюдается отрицательный эффект (NOAEL), факторов, которые признаны критическими, и применение нескольких поправочных коэффициентов для учета различных неопределенных условий.
Требования к данным для установления опасности
Установление опасности представляет собой качественную оценку тех свойств субстанции, которые вызывают отрицательные эффекты. При установлении опасности следует рассмотреть все доступные данные для животных и человека для каждого со- единения. Данные для установления опасности должны включать неклинические фармакодинамические данные, исследования токсичности повторных доз, исследования канцерогенности, исследования генотоксичности in vitro и in vivo, исследования токсичности на репродуктивные свойств и развития, а также клинические данные (терапевтические и отрицательные эффекты). Отсутствие данных для активных субстанций варьируется в зависимости от стадии разработки и симптомов. Если наборы данных не полны, то следует критически оценить идентифицированные пробелы с учетом влияния, которое они могут оказать на получение надежных величин пределов воздействия на здоровье.Установление критических эффектов
К критическим эффектам относятся наиболее чувствительные индикаторы отрицательного эффекта, наблюдаемого при доклинических исследованиях токсичности, если только не приведено ясное свидетельство (например, из исследования механизма действия препарата, фармакодинамических данных и т. д.), что такие наблюдения не относятся к человеку или целевому животному. К критическим эффектам относится также любой клинический терапевтический и отрицательный эффекты.Установление величины NOAEL
Следует установить величину NOAEL для всех критических эффектов. Величина NOAEL представляет собой наибольшую испытанную дозу, при которой не наблюдается «критический» эффект. Если критический эффект наблюдается в нескольких исследованиях на животных, то величину NOAEL для наименьшей дозы следует использовать для вычисления PDE. Если отсутствуют данные по NOAEL, то может использоваться уровень, при котором наблюдается наименьший отрицательный эффект. Вели- чина NOAEL, основанная на клинических фармакодинамических эффектах, должна соответствовать наибольшей испытанной дозе, которая считается терапевтически неэффективной.Применение поправочных коэффициентов
Величина PDE вычисляется путем деления NOAEL для критического эффекта на различные поправочные коэффициенты (также именуемые коэффициентами безопасности, неопределенности, оценки или модификации) для учета различных неопределенностей и экстраполяции до надежного и устойчивого уровня отсутствия эффекта для человека или популяции целевых животных. Коэффициенты F1 – F5 относятся к следующим видам неопределенности: F1 – коэффициент (значения от 1 до 12) для учета экстраполяции между видами; F2 – коэффициент, равный 10, для учета различий между индивидуумами; F3 – коэффициент, равный 10, для учета исследований токсикологических свойств в течение короткого периода времени, т.е. меньше 4 недель; F4 – коэффициент (значения от 1 до 10), применяемый в случаях серьезной токсичности, например, генотоксической канцерогенности, нейротоксичности или тератогенности; F5 – переменный коэффициент, который может использоваться, если не установлен уровень, при котором эффект не наблюдается. Если известно только значение LOEL, то может использоваться коэффициент до 10 в зависимости от тяжести токсичности. Могут использоваться дополнительные поправочные коэффициенты для учета неопределенностей по остаткам неучтенных выше рассмотренными коэффициентами, если они основаны на опубликованных данных и проведено достаточное обсуждение, например, недостаточность данных по токсическому влиянию на репродуктивные свойства и развитие (см. раздел 5.4). Дальнейшие указания по выбору поправочных коэффициентов F1 и F4 даны в приложениях 3 к Руководствам ICH Q3C (R4) и VICH GL 18. Использование и выбор поправочных коэффициентов должны быть обоснованы. Допускается ограничение на использование коэффициентов F2 и, возможно, F5 при получении PDE на основе окончательных данных для человека. Допускаются отклонения от стандартных значений коэффициентов, приведенных выше, при наличии адекватного и научного обоснования.Выбор окончательного значения PDE
Если при вычислении более чем одной величины PDE установлены несколько критических эффектов, то следует принять наиболее подходящий PDE для валидации процесса очистки с обоснованием. Как правило, используется наименьшее значение PDE.4.2 Использование клинических данных
Целью определения пределов воздействия на здоровье является обеспечение безопасности человека, в связи с чем предъявляются высокие требования к качеству клинических данных для человека. Не предусмотренные фармакодинамические эффекты у пациентов, вызванные загрязнением активных субстанций, могут привести к опасности, поэтому при установлении критического эффекта следует учитывать кинические фармакологические данные. Следует принимать во внимание, до какой степени рассматриваемая активная субстанция связана с критическими отрицательными эффектами при клинических испытаниях. Если наиболее критический эффект, установленный для определения предела воздействия на здоровье, основан на фармакологических и/или токсикологических эффектах, наблюдавшихся на людях, а не на животных, то использование формулы для определения PDE может быть неправомерным и для этой цели может использоваться оценка клинических данных для специфической субстанции.4.3 Экстраполяция для других путей введения
Величина PDE, получаемая для активных субстанций (загрязнителя), обычно основана на исследованиях для определенного клинического пути введения. Но возможно последовательное производство активных субстанций или лекарственного средства с различными путями введения на одном и том же оборудовании (в помещении). Изменение пути введения может привести к изменению биодоступности. В связи с этим следует применять поправочные коэффициенты для экстраполяции между путями введения, если есть четкое различие (например, > 40 %) по отношению к биодоступности для заданного пути введения. Поскольку биодоступность может различаться между видами (потребителями), то поправочные коэффициенты для экстраполяции для других путей введения должна основываться преимущественно на данных для человека или в случае ветеринарных лекарственных средств на данных для целевого животного. В случае, если данные о биодоступности для человека или целевого животного для разных путей введения неизвестны и предполагается, что изменение пути введения может привести к увеличению систематического введения загрязнителя (например, при переходе от орального применения к ингаляции), может быть выполнена консервативная экстраполяция с допущением 100%-ой биодоступности загрязнителя. На- пример, в случае экстраполяции оральное введение – ингаляция величина PDE, полученная для орального введения, может быть откорректирована путем умножения на следующий поправочный коэффициент: Поправочный коэффициент (оральное введение – ингаляция): % абсорбции при оральном введении/100% респираторной абсорбции В случаях, если данные о биодоступности для человека или целевого животного для других путей введения неизвестны и если может предполагаться, что систематическое введение загрязнителя будет меньше, чем для загрязненной активной субстанции/лекарственного средства, то необходимость применения поправочного коэффициента при вычислениях PDE отсутствует. Предполагается, что экстраполяция для других путей введения будет выполняться в индивидуальном порядке.5. Особые условия
5.1 Активные субстанции с генотоксичным действием
Для генотоксичных субстанций, у которых нет явных различий в порогах, считается, что любой уровень их введения связан с риском. Однако для генотоксичных веществ, для которых не установлен порог, предварительный уровень допустимого рис- ка установлен руководством EMA Guideline on the Limits of Genotoxic Impurities (Руководство по пределам генотоксичных загрязнений) в виде предела токсикологического значимости (Threshold of Toxicological Concern, TTC) 1,5 мкг/человек/день. Если вели- чина ТТС представляет уровень введения генотоксичного загрязнения, связанный с теоретическим риском заболевания раком как один случай дополнительного заболевания раком на 100 000 пациентов при введении (получении) в течение всей жизни. Принимая тот факт, что продолжительность получения остатков активной субстанции будет существенно более ограниченной (например, потому что на практике уровни переносимых остатков активной субстанции могут снижаться от серии к серии), пределы, основанные на максимальных данных, в этом случае получение 1,5 мкг/человек/день не будет превышать теоретический риск заболевания раком 1х10-6. Следовательно, для остатков активных субстанций с неустановленным пределом может быть применена предельная доза 1,5 мкг/человек/день. Для ветеринарного препарата, который может быть загрязнен активной субстанцией, следует использовать ту же величину ТТС. Но для показателя «на кг веса тела» (т. е. ТТС равно 0,03 мкг/кг веса тела/день). Если загрязненный продукт вводится животным, предназначенным для употребления в пищу, то предел переноса должен учитывать условия безопасности как для целевого животного, так и для потребителя. По- этому следует показать, основываясь на худшем случае сценария введения, что ни целевое животное, ни потребитель не получат остатки активной субстанции при уровнях, превышающих ТТС. Если для генотоксичных активных субстанций есть удовлетворительные данные по канцерогенности, то следует применить оценку риска индивидуально для данного соединения, чтобы получить допустимую дозу приема вместо применения критерия ТТС. Если для генотоксичных фармацевтических субстанций есть удовлетворительные данные по механизму действия препарата, связанному с пределом, то безопасные уровни введения без риска по генотоксичности могут быть установлены на основе подхода PDE.5.2 Активные субстанции с высоким сенсибилизирующим потенциалом
Реакции гиперчувствительности при введении иммунных лекарственных средств могут проявляться у чувствительных пациентов. Наблюдаемые реакции могут варьироваться от мягких случаев контактной чувствительности до потенциально летальных анафилактических реакций. Как указано в п. 3.6 главы 3 части I правил GMP, следует предусматривать выделенное оборудование (помещения) для производства активных субстанций и лекарственных средств с высоким сенсибилизирующим потенциалом, для которых отсутствуют научные данные по допустимым уровням воздействия, или риск при обращении с продуктом на оборудовании (помещениях) не может адекватно контролироваться путем организационных или технических мер. Классификация активной субстанции или лекарственного средства с высоким сенсибилизирующим потенциалом должна учитывать, насколько частыми являются проявления чувствительности к субстанции у людей; или вероятность высокой чувствительности у людей, основываясь на данных о животных или других валидированных тестах. Следует также учитывать тяжесть этих реакций и учитывать ее при оценке.5.3 Терапевтические макромолекулы и пептиды
Известно, что терапевтические макромолекулы и пептиды могут деградировать и денатурировать при воздействии веществ с экстремальными значениями рН и/или тепла и могут утратить фармакологическую активность. Очистка биофармацевтического технологического оборудования обычно выполняется при воздействии на поверхности оборудования веществ с экстремальными значениями рН и/или тепла, которые приводят к разрушению и инактивации продуктов на основе белков. В связи с этим может не потребоваться определение пределов воздействия на здоровье с использованием пределов PDE для активной и неповрежденной продукции. Если возможны другие пути перекрестных загрязнений, то следует учитывать риск в индивидуальном порядке.5.4 Недостаточность данных по токсическому влиянию на репродуктивную функцию и развитие для животных
Для обеспечения защиты всех популяций следует снижать остатки активных субстанций до уровня, который не представляет риск для репродуктивной функции и развития. Однако на ранних стадиях разработки могут отсутствовать неклинические данные для оценки токсического влияния на репродуктивную функцию и развитие новых активных субстанций. Для разрешенных для применения лекарственных средств могут также иметь место пробелы в научных знаниях, например, свойство специфических для самцов препаратов вызывать отрицательные эффекты в развитии эмбрионов. В этих случаях может использоваться величина NOAEL на основе длительных исследований в вычислении PDE с применением дополнительного поправочного коэффициента (например, 10) при должном обосновании. Если известны данные исследований по токсическому влиянию на репродуктивную функцию и развитие для близких соединений, то могут использоваться специфические для класса данные для установления опасности неиспытанных загрязнителей путем сплошного анализа.5.5 Лекарственные средства для исследований
Оценка величины PDE путем установления пределов может быть затруднена на ранних стадиях разработки лекарственных средств для исследований (стадии I/II). В этих случаях для получения пределов воздействия на здоровье может использоваться альтернативный подход с классификацией по специфическим пределам для классов, например, на основе низкой/высокой ожидаемой фармакологической активности, низ- кой/высокой токсичности, генотоксичности/канцерогенности, аналогично многоуровневым порогам токсикологической оценки (ТТС) [1-3], при должном обосновании. Поскольку большинство допустимых пределов определено при длительном воздействии, то верхний предел может быть обоснован, если субстанция выпускается на одном оборудовании с другой субстанцией, предназначенной для испытаний в течение короткого времени [4]. При наличии более подробных фармакологических и токсикологических данных следует вычислять специфические для данного соединения пределы, как указано выше для пределов воздействия на здоровье.6. Отчет о порядке определения PDE
Идентификация «критических эффектов» при установлении PDE согласно разделу 4 должна основываться на полном исследовании литературы, включая справочники и монографии, а также исследований научных баз данных в электронной форме. Порядок исследований и полученные результаты должны быть представлены в четкой документальной форме. После рассмотрения экспертом компания должна организовать дискуссию с учетом критических факторов и обоснования для выбора факторов и дозы, которые должны быть использованы при получении PDE. Следует дать ссылки на основные исследования на животных и человеке, используемые для получения вели- чины PDE; эти исследования должны быть рассмотрены с учетом их качества (методика исследований, описание результатов, тщательность отчета). Подход к определению PDE должен обеспечить четкое обоснование поправочным коэффициентам, которые применяются при определении PDE. Более того, для представления обзора инспектору GMP первая страница документа по подходу к определению PDE должна со- держать основные данные о процессе оценки (пример приведен в приложении).7. Внедрение
Данное руководство предлагает метод идентификации риска для применения в основанных на научных данных и анализе риска подходах к производству лекарственных средств на одном и том же технологическом оборудовании (помещении) в соответствии с главами 3 и 5 части I правил GMP. Чтобы позволить производителям ввести в действие данное руководство, установлены следующие сроки: Для лекарственных средств, намеченных к производству впервые на одном и том же оборудовании (помещении): 6 мес. После публикации данного руководства; Для уже выпускаемых лекарственных средств на одном и том же оборудовании (помещении) руководство вступает в силу или существующие меру должны быть обоснованы в течение:- Одного года после публикации данного руководства для производите- лей продукции для человека, включая производителей лекарственных средств для человека и животных с использованием одного и того же оборудования (помещений);
- Двух лет после публикации данного руководства для производителей, выпускающих продукцию только для ветеринарии.
8. Определения
| F: | Поправочный коэффициент |
| GMP: | Правила производства лекарственных средств |
| ICH: | Международная конференция по гармонизации |
| LOAEL: | Уровень, при котором наблюдается наименьший отрицательный эффект |
| PDE: | Допустимое дневное воздействие |
| ADE: | Разрешаемое дневное воздействие |
| NOAEL: | Уровень, при котором не наблюдается отрицательный эффект |
| TTC: | Предел токсикологического воздействия |
| VICH | Ветеринарная международная конференция по гармонизации |
- Kroes R, Renwick A, Cheeseman M, Kleiner J, Mangelsdorf I, Piersma A, Schilter B, Schatter J, van Schothorst F, Vos JG, Würtzen G. (2004). Structure-based thresholds of toxicological concern (TTC): guidance for application to substances present at low levels in the diet. Fd Chem Toxicol 42, 65-83.
- Munro IC, Renwick AG, Danielewska-Nikiel B (2008). The threshold of toxicologi- cal concern (TTC) in risk assessment. Toxicol Lett 180, 151-156.
- Dolan DG, Naumann BD, Sargent EV, Maier A, Dourson M (2005). Application of the threshold of toxicological concern concept to pharmaceutical manufacturing operations. Regul Toxicol Pharmacol, 43, 1-9.
- Bercu JP & Dolan DG, (2013). Application of the threshold of toxicological concern concept when applied to pharmaceutical manufacturing operations intended for short-term clinical trials. Regul Toxicol Pharmacol. 2013 Feb;65(1):162-7.
Приложение
Порядок определения величины PDE
Наименование компании Адрес компании Имя и подпись эксперта Дата Дата рассмотрения Наименование химического вещества Установленная опасность ДА НЕТ НЕИЗВЕСТНООснова для определения PDE
Обоснование выбора «основного» критического эффекта, используемого для вы- числения NOAEL и поправочные коэффициенты для определения PDEСсылки
Публикации, использованные для идентификации критического эффекта и дозы.Данные об эксперте
Приложение
Головка бланка письма производителяПисьменное подтверждение
для активных субстанций для лекарственных средств для человека, экспортируемых в Европейский Союз, в соответствии со статьей 46b(2)(b) Директивы 2001/83/ЕС Подтверждение № (присваивается надзорным органом, выдающим подтвер- ждение):- Наименование адрес предприятия (включая номер строения, где требуется):
- Номер(а) лицензий производителя:1
| Активная субстанция(и):2 | Вид(ы) деятельности:3 |
Общая часть
Содержание Сертификата на серию для лекарственных средств
(Примечание: См. Пояснительную записку и словарь для обеспечения единства терминов) [Головка бланка письма производителя-экспортера]- Наименование продукции
- Страна-импортер
- Номер лицензии на реализацию или номер лицензии на проведение клинических испытаний
- Эффективность/активность
- Дозированная форма
- Размер и вид упаковки
- Номер серии
- Дата изготовления
- Срок годности
- Наименования, адреса и номера лицензий всех производственных площадок и организаций (подразделений) по контролю качества
- Сертификаты соответствия GMP всех площадок, указанных в п. 10 или ссылочные номера регистра GMP EC (EudraGMP)
- Результаты анализа
- Комментарии.
- Заключение о сертификации
- Имя, должность и титул лица, выдавшего разрешение на выпуск
- Подпись лица, выдавшего разрешение на выпуск
- Дата подписи
Пояснительная записка и словарь
1. Наименование продукции
Вид собственности, фирменный знак или торговая марка или точное наименование в стране-импортере, как принято. Для лекарственных средств, предназначенных для исследований (IMP), номер кода в соответствии с заявкой на проведение клинических испытаний.2. Страна-импортер
3. Номер лицензии на реализацию или номер лицензии на проведение клинических испытаний
Номер лицензии на реализацию продукта в стране-импортере. Для IMP указывается ссылочный номер лицензии на проведение клинических испытаний, при наличии.4. Эффективность/активность
Идентификационное обозначение (наименование) и количество в дозированной форме для всех активных ингредиентов (составляющих). Препараты для клинических исследований (IMPs), включают плацебо, для них указывается способ нанесения информации, которая не должна быть раскрывать исследования.5. Дозированная форма или фармацевтическая форма, например, таблетки, капсулы, мази и др.
6. Размер и вид упаковки
Приводится вид упаковки и флаконов, бутылок, блистеров и др.7. Номер серии
или номер партии продукции. Указывается неповторимая комбинация цифр, букв или символов, которая идентифицирует серию и по которой могут быть определены продукт и его история.8. Дата изготовления
Указывается в соответствии национальными (местными) требованиями страны- импортера.9. Срок годности
На контейнере с продуктом (первичной упаковке) или этикетке указывается дата, обозначающая время, в течение которого предполагается, что срок хранения, согласован- ный страной-импортером, не истечет при хранении в заданных условиях и по истечении которого использование продукта не допускается.10. Наименования, адреса и номера лицензий всех производственных площадок и организаций (подразделений) по контролю качества
Приводятся все площадки (подразделения, организации), причастные к производству (включая упаковку, маркировку и контроль качества серии) с указанием их наименований, адресов, номеров лицензий. Наименования и адреса должны совпадать с указанными в лицензиях на производство.11. Сертификаты соответствия GMP всех площадок, указанных в п. 10 или ссылочные номера регистра GMP EC (EudraGMP)
Указываются номера сертификатов и/или ссылочные номера в реестре GMP EC (EudraGMP).12. Результаты анализа
Приводятся утвержденные спецификации, все полученные результаты и ссылки на использованные методы (могут быть приведены ссылки на отдельные сертификаты анализа, в которых должны быть указаны даты и поставлены подписи).13. Комментарии/замечания
Приводятся любые дополнительные данные, которые могут представить интерес для импортера и/или инспектора, проверяющего соответствие сертификата на серию (например, специальные условия хранения и транспортирования).14. Заключение о сертификации
Это заключение должно содержать данные о производстве, включая упаковку, маркировку и контроль качества. Рекомендуется использовать следующую форму текста: «Настоящим я удостоверяю, что приведенные выше данные верны и точны. Эта серия продукции произведена (включая упаковку, маркировку и контроль качества) на указан- ной выше площадке (площадках) в полном соответствии с требованиями местных надзорных органов по GMP, которые указаны в лицензии на реализацию страны-импортера или спецификации на продукт для лекарственных средств, предназначенных для исследований. Протоколы производства, упаковки и анализа (контроля) рассмотрены и признаны соответствующими требованиям GMP».15. Имя, должность и титул лица, выдавшего разрешение на выпуск
Приводятся имена и адреса, если указывается более чем одна площадка по п. 10.16. Подпись лица, выдавшего разрешение на выпуск
17. Дата подписи
Словарь эквивалентных терминов в форме сертификата (не является исчерпывающим)
Активные субстанции (active substances) = активные фармацевтические ингредиенты/составляющие (active pharmaceutical ingredients/constituents) Серия (batch) = партия (lot) Дозированная форма (dosage form) = фармацевтическая форма (pharmaceutical form) Производитель (manufacture) = изготовитель (fabricator) Производство (manufacturing/manufacture) = изготовление (fabrication) Лицензия на производство (manufacturing authorisation) = (establishment licence) Лекарственное средство (medicinal product) = фармацевтическое средство (pharma-ceutical product) = препарат (drug product) Контроль качества (quality control) = испытания (testing)1.Фармацевтическая система качества
1.1 Введение
Данный документа представляет собой трехсторонне руководство ICH, описывающее модель эффективной системы обеспечения качества для фармацевтической промышленности, определяемой как Фармацевтическая система качества. В тексте всего документа термин «фармацевтическая система качества» относится к ICH Q10. ICH Q10 содержит описание всесторонней модели эффективной фармацевтической системы качества, которая основана на концепциях качества Международной организации по стандартизации (ИСО), руководствах GMP (Правила производства лекарственных средств) и дополняется руководствами ICH Q8 «Разработка фармацевтической продукции» и ICH Q9 «Анализ рисков». ICH Q10 представляет собой модель фармацевтической системы качества, которая может применяться в течение всего жизненного цикла продукции. Многое из содержания ICH Q10, применимого к производственным площадкам, содержится в региональных требованиях GMP. Поэтому содержание ICH Q10 является дополнением для добровольного применения (опцией) к действующим региональным требованиям GMP. ICH Q10 демонстрирует поддержку эффективной фармацевтической системы качества промышленностью и надзорными органами для повышения качества и доступности лекарственных средств в мировом масштабе интересах здоровья населения. Применение ICH Q10 в течение всего жизненного цикла продукции должно способствовать инновациям и постоянному улучшению фармацевтического производства и усилению связи между разработкой фармацевтической продукции и производством.1.2. Область применения
Данное руководство относится к системам, поддерживающим разработку и производство субстанций лекарственных средств (например, активных фармацевтических субстанций, АФС), лекарственных средств, включая биотехнологические и биологические продукты, в течение всего жизненного цикла. Элементы руководства ICH Q10 следует применять в соответствии и в степени, необходимой для каждого этапа жизненного цикла, учитывая различия между этапами и их назначение (раздел 3). Для целей данного руководства жизненный цикл продукции включает в себя следующие технические действия для новых и существующих видов продукции:- Фармацевтическая разработка;
- Разработка субстанции лекарственного средства;
- Разработка лекарственной формы (включая вид первичной упаковки и укупорки);
- Производство продукции для исследований;
- Разработка системы реализации (где требуется);
- Разработка технологического процесса и увеличение объемов выпуска (масштабирование);
- Разработка аналитических методов.
- Передача (освоение) технологии:
- Передача нового продукта в процессе разработки и производства;
- Передача внутри или между производственными площадками и контрольными подразделениями для реализуемых продуктов.
- Коммерческое производство:
- Приобретение и контроль материалов;
- Обеспечение помещениями, инженерными системами и оборудованием;
- Производство (включая упаковку и маркировку);
- Контроль и обеспечение качества;
- Выпуск;
- Хранение;
- Реализация (включая оптовую торговлю).
- Прекращение выпуска продукции:
- Хранение документации;
- Хранение образцов;
- Непрерывная оценка продукции с документальным оформлением.
1.3. Связь ICH Q10 с региональными требованиями GMP, стандартами ИСО и ICH Q7
Региональные руководства по GMP, руководство ICH Q7 «Правила GMP для производства активных фармацевтических субстанций» и стандарты ИСО по системе качества являются основой руководства ICH Q10. Для достижения излагаемых ниже целей ICH Q10 дополняет требования GMP описанием специфических элементов системы качества и ответственности руководителей. ICH Q10 дает гармонизированную модель для фармацевтической системы качества на протяжении всего жизненного цикла продукта и направлено на применение совместно с региональными требованиями GMP. Региональные требования GMP не обязательно включают все стадии жизненного цикла (например, разработку). Элементы системы качества и ответственность руководителей, рассматриваемые в данном документе, направлены на усиление использования науки и анализа рисков в применении к каждому этапу жизненного цикла, способствуя, таким образом, постоянному улучшению на протяжении всего жизненного цикла.1.4. Связь ICH Q10 с требованиями надзорных органов
Подход надзорных органов к отдельному продукту или производству должен соответствовать уровню понимания продукта или процесса, результатам оценки риска и эффективной системы обеспечения качества. После внедрения фармацевтической системы качества ее эффективность, как правило, может быть оценена при инспектировании производственной площадки надзорным органом. Потенциальные возможности для работы надзорных органов на основе усовершенствованного научного подхода и анализа рисков определены в приложении 1. Порядок инспектирования устанавливается на уровне регионе.1.5. Цели ICH Q10
В результате применения модели Q10 ожидается достижение трех основных целей, которые дополняют или усиливают региональные требования GMP.1.5.1. Выпуск продукции
Следует определить, внедрить и поддерживать систему, которая позволяет выпускать продукцию с показателями качества, необходимыми для удовлетворения нужд пациентов, специалистов в области здравоохранения, надзорных органов (включая соответствие утвержденным требованиям) и других внутренних и внешних потребителей.1.5.2. Определение и поддержание состояния контроля
С целью обеспечения постоянного соответствия и требуемых возможностей процесса следует разработать и применять эффективную систему контроля и регулирования. При организации такой системы может быть полезным анализ рисков.1.5.3. Организация постоянного улучшения (совершенствования)
Следует определить и реализовать меры по улучшению качества продукции, совершенствованию процесса, уменьшению изменчивости, внедрению инноваций и совершенствованию фармацевтической системы качества и на этой основе обеспечить постоянное повышение качества. Анализ рисков может быть полезным при выборе зон для постоянного улучшения и выбора приоритетов.1.6. Средства достижения: обеспечение информацией и анализ рисков
Обеспечение информацией персонала и анализ рисков для качества позволяют компании внедрять ICH Q10 эффективно и успешно. Эти средства обеспечивают достижение целей, приведенных в разделе 1.5 (см. выше), за счет применения решений, основанных на научном подходе и анализе, относящихся к качеству продукции.1.6.1. Обеспечение информацией
Следует совершенствовать знания о продукте и процессе от его разработки и коммерческого производства до прекращения выпуска продукции. Например, разработка с использованием научных подходов дает знания для понимания продукта и процесса. Обеспечение информацией представляет собой систематический процесс приобретения, анализа, хранения и распространения информации о продукции, процессах производства и составных частях. К источникам знаний относятся предшествовавшие знания (полученные в общедоступной системе или на предприятии с документальным подтверждением), исследования по фармацевтической разработке, работы при передаче технологии, исследования при аттестации (валидации) в течение всего жизненного цикла, производственный опыт, внедрение нового, постоянное совершенствование, внесение изменений и т. д. (перечень не исчерпывающий).1.6.2. Анализ рисков
Анализ рисков является составной частью фармацевтической системы качества. Он может включать в себя предупреждающие действия по установлению, научнооценке и недопущению возможных рисков для качества. Он способствует постоянному улучшению процессов и качества продукции в течение жизненного цикла. ICH Q9 устанавливает принципы и дает примеры использования методов анализа рисков, которые могут применяться к разным сторонам фармацевтического качества.1.7. Требования к построению и содержанию
- Построение, организация и документация фармацевтической системы качества должны иметь четкую структуру и быть ясными для понимания и применения.
- Элементы ICH Q10 следует применять в соответствии и в степени, необходимой для каждого этапа жизненного цикла, с учетом специфики каждого этапа и знаний о нем.
- При разработке новой фармацевтической системы качества или изменении существующей следует учесть размеры и сложность работы компании. Структура фармацевтической системы качества должна включать в себя соответствующие методы анализа рисков. Некоторые аспекты фармацевтической системы качества могут относится ко всей компании, а другие – только к определенному производству. Эффективность фармацевтической системы качества обычно оценивается на уровне производства.
- Фармацевтическая система качества должна включать в себя необходимые процессы, ресурсы и ответственность, чтобы обеспечить качество работ по контракту и качество приобретаемых материалов в соответствии с п. 7.
- Ответственность руководителей согласно разделу 2 должна быть определена в фармацевтической системе качества.
- Фармацевтическая система качества должна включать в себя следующие элементы (раздел 3): описание технологического процесса и порядок контроля качества продукции, корректирующие и предупреждающие действия, порядок контроля изменений и анализ работы.
- Следует определить и использовать показатели для контроля эффективности процессов в рамках фармацевтической системы качества (раздел 4).
1.8. Руководство по качеству
Следует разработать руководство по качеству или эквивалентный документ с описанием фармацевтической системы качества. Это описание должно включать в себя:- Политику в области качества (раздел 2);
- Область применения фармацевтической системы качества;
- Перечень процессов фармацевтической системы качества в требуемой последовательности, связи и взаимозависимости. Схемы процессов и диаграммы потоков могут быть полезными средствами для наглядного представления фармацевтической системы качества;
- Ответственность руководителей в рамках фармацевтической системы качества (раздел 2).
2. Ответственность руководства
Руководство играет важную роль в формировании и выполнении обязательств компании в области качества и работе фармацевтической системы качества2.1. Обязанности руководства
- Высшее руководство несет исключительную ответственность за обеспечение эффективности фармацевтической системы качества на предприятии с целью достижения целей качества и за распределение функций, обязанностей и ответственности, организацию взаимодействия и внедрения на всем предприятии.
- Руководители обязаны:
- Участвовать в разработке, внедрении, контроле и поддержании эффективной фармацевтической системы качества;
- Оказывать энергичную и зримую поддержку фармацевтической системе качества и обеспечить ее внедрение на всем предприятии;
- Своевременно организовать эффективное взаимодействие и улучшение существующих процессов для повышения качества на соответствующем уровне управления;
- Определять индивидуальные и коллективные обязанности, ответственность и права и взаимодействие всех элементов организационной структуры, относящихся к фармацевтической системе качества. Обеспечить работу и понимание этого взаимодействия на всех уровнях организации. Местные надзорные органы требуют выделения независимого подразделения (структуры) качества для выполнения определенных обязанностей в фармацевтической системе качества;
- Проводить рассмотрение процесса и качества продукции и фармацевтической системы качества;
- Способствовать постоянному улучшению;
- Привлекать необходимые ресурсы.
2.2. Политика в области качества
- Высшее руководство должно определить политику в области качества, которая отражает общие намерения и направление работы компании в области качества.
- Политика в области качества должна включать установку на выполнение требований надзорных органов и должна нацеливать на постоянное улучшение фармацевтической системы качества.
- Политика в области качества должна быть воспринята и понята персоналом на всех уровнях компании.
- Следует периодически рассматривать политику в области качества для обеспечения ее постоянной эффективности.
2.3. Планирование качества
- Высшее руководство должно обеспечить постановку и решение задач, необходимых для реализации политики в области качества.
- Эта работа должна быть поддержана всех причастных уровнях компании.
- Эта работа должна соответствовать стратегии компании и политике в области качества.
- Руководство должно обеспечить ресурсами достижение поставленных целей в области качества и организовать обучение.
- Следует определить показатели работы, которые отражают прогресс в реализации политики в области качества, регулярно контролировать их выполнение в соответствии с п. 4.1. данного документа.
2.4. Обеспечение ресурсами
- Руководство должно обеспечить работу необходимыми ресурсами (людскими, финансовыми, материальными, оборудованием и помещениями) для достижения целей фармацевтической системы качества и постоянного повышения ее эффективности.
- Руководство должно обеспечить соответствие ресурсов конкретному продукту, процессу и площадке.
2.5. Внутренне связи
- Руководство должно обеспечить создание и внедрение взаимосвязей внутри организации.
- Взаимосвязи должны обеспечивать обмен информацией между всеми уровнями компании.
- Взаимосвязи должны обеспечивать необходимое и своевременное повышение качества конкретного продукта и аспектов, связанных с фармацевтической системой качества.
2.6. Рассмотрение руководством
- Высшее руководство несет ответственность за управление фармацевтической системой качества путем рассмотрения работы для обеспечения постоянного соответствия и эффективности.
- Руководство должно оценивать выводы периодических анализов функционирования процесса и качества продукции и фармацевтической системы качества в соответствии с разделами 3 и
2.7. Управление работами, выполняемыми по контракту и работа в приобретаемыми материалами
Фармацевтическая система качества, включая ответственность руководства согласно данному разделу, распространяется на контроль и анализ любой деятельности по контрактам и качества приобретаемых материалов. Фармацевтическая компания несет исключительную ответственность за организацию порядка контроля работ по контрактам и качество приобретаемых материалов. Этот порядок должен включать анализ рисков и следующие требования:- Предварительную оценку выполнения работ по контракту или выбора поставщиков материалов, пригодности и компетентности другой стороны для выполнения взятых на себя обязательств или обеспечения материалами по определенной цепи поставок (например, путем аудитов, оценки материалов, аттестации);
- Определение ответственностей и порядки взаимодействия с причастными сторонами для действий, связанных с качеством. Для работ по контракту это должно включать письменное соглашение между заказчиком и исполнителем;
- Контроль и анализ выполнения обязательств исполнителем или качества материалов от поставщика и идентификацию и внедрение любых требуемых мер по улучшению;
- Входной контроль ингредиентов и материалов для проверки получения их от утвержденного источника по согласованной цепи поставок.
2.8. Изменения в праве собственности на продукцию
При изменении права собственности (например, при приобретении) следует учитывать сложность этого процесса и обеспечивать:- Установление ответственности для каждой причастной стороны;
- Передачу необходимой информации.
3. Постоянное улучшение процесса и качества продукции
В данном разделе дано описание целей стадий жизненного цикла и четырех специфических для фармацевтической системы качества элементов, которые усиливают региональные требования по достижению установок ICH Q10, как указано в п. 1.5. Это не заменяет все региональные требования.3.1. Цели этапов жизненного цикла
Ниже приводится описание целей каждого этапа жизненного цикла продукции.3.1.1. Фармацевтическая разработка
Целью фармацевтической разработки является создание продукта и технологического процесса производства для постоянного достижения ожидаемого результата и удовлетворения нужд пациентов, специалистов в области здравоохранения, надзорных органов и внутренних требований потребителей. Подходы к фармацевтической разработке даны в руководстве ICH Q8. Результаты исследований и клинических испытаний не входят в область применения данного руководства, но вносят вклад в фармацевтическую разработку.3.1.2. Передача технологии
Целью передачи технологии является передача продукта и знаний о процессе от стадии разработки в производство и внутри или между производственными площадками для выпуска продукции. Знания дают основу для технологического процесса, стратегии контроля, подходов к аттестации и последующему постоянному улучшению.3.1.3. Коммерческое производство
К целям производства относятся выпуск продукции, обеспечение контроля и постоянное улучшение. Фармацевтическая система качества должна обеспечивать требуемое качество продукции в производстве, выполнение требований к процессу и контролю, возможности определения и оценки улучшения и постоянное расширение объема знаний.3.1.4. Прекращение производства продукции
При прекращении производства продукции следует обеспечить эффективное управление завершающим этапом жизненного цикла продукции. При прекращении производства продукции следует выполнить ряд запланированных мероприятий, таких как организация хранения документации, образцов и постоянная оценка продукции (например, рекламаций и контроль стабильности) и извещение надзорных органов в установленном порядке.3.2. Элементы фармацевтической системы качества
Приводимые ниже элементы могут входить в региональные требования GMP. Но модель Q10 направлена на усиление этих элементов с целью продвижения подхода жизненного цикла к качеству продукции. К этим четырем элементам относятся:- Система контроля процесса и качества продукции;
- Система корректирующих и предупреждающих действий (Corrective action and preventive action – CAPA);
- Система внесения изменений;
- Анализ процесса и качества продукции.
3.2.1. Система контроля процесса и качества продукции
Фармацевтическим компаниям следует создавать и применять систему контроля процесса и качества продукции для повышения статуса контролируемости производства. Эффективная система контроля дает уверенность в постоянной способности процесса и системы контроля выпускать продукцию требуемого качества и обозначить зоны для постоянного улучшения. Система контроля процесса и качества продукции должна:- Применять анализ рисков для определения стратегии контроля. К этому могут относиться параметры и атрибуты, относящиеся к субстанциям лекарственных средств, материалам и компонентам для лекарственных средств, помещениям и оборудованию, условиям эксплуатации, внутрипроизводственному контролю, спецификациям на готовую продукцию, соответствующим методам и периодичности контроля. Стратегия контроля должна способствовать своевременной обратной связи/предпринимаемым действиям и обеспечивать соответствующие корректирующие и предупреждающие действия;
- Обеспечить приборами для анализа и контроля параметров и свойств, указанных в стратегии контроля (например, регистрация данных и статистические методы);
- Выполнить анализ параметров и свойств, указанных в стратегии контроля, для сверки выполняющегося процесса с параметрами контроля;
- Установить источники отклонений, влияющих на процесс и качество продукции, для возможного устойчивого уменьшения отклонений или установления контроля над ними;
- Установить обратную связь в отношении качества продукции как от внутренних, так и от внешних источников, например, рекламаций, отклонений продукции, несоответствий, отзывов, девиаций, аудитов и инспекций надзорных органов и их результатов;
- Обеспечить повышение квалификации для лучшего понимания процесса, расширения параметров для разработки (где они установлены) и способствовать применению новых подходов к аттестации (валидации) процессов.
| Фармацевтическая разработка | Передача технологии | Коммерческое производство | Прекращение производства |
| Полученные знания о процессе и продукте и контроль продукта в течение этапа разработки могут использоваться для установления стратегии контроля при производстве | Контроль при увеличении объема производства может дать предварительную оценку процесса и быть успешно интегрированным в производство. Знания, полученные при передаче технологии и увеличении объема производства, могут быть полезными при дальнейшей разработке стратегии контроля | Следует применять хорошо определенную систему контроля процесса и качества продукции для обеспечения работы в контролируемом состоянии и определения зон для улучшения | После прекращения производства следует продолжать контроль, такой как исследование стабильности до его завершения. Следует продолжать контроль за выпущенной на рынок продукцией в соответствии с региональными требованиями |
3.2.2. Система корректирующих и предупреждающих действий (САРА)
Фармацевтическая компания должна иметь систему корректирующих и предупреждающих действий на основе анализа рекламаций, отклонений продукции, несоответствий, отзывов, девиаций, аудитов и инспекций надзорных органов и их результатов, а также анализа тенденций при выполнении процесса и контроле качества продукции. Для установления первопричины при проведении анализа следует использовать структурный подход. Затраты труда, степень формализации и документального оформления при проведении анализа должны соответствовать степени риска с учетом ICH Q9. Методология CAPA должна привести к улучшению продукции и процесса и повысить уровень их понимания. Таблица 2. Применение системы корректирующих и предупреждающих действий в течение жизненного цикла продукта| Фармацевтическая разработка | Передача технологии | Коммерческое производство | Прекращение производства |
| Проводится анализ отклонений в процессе или продукте. Методология САРА полезна при сочетании корректирующих и предупреждающих действий с итеративным процессом разработки и развития | САРА может использоваться в качестве эффективной системы обратной связи, обоснования предпринимаемых действий и постоянного улучшения | Следует использовать САРА и оценивать эффективность принятых мер | Следует продолжать САРА после прекращения производства. Следует учитывать влияние на продукцию, оставшуюся на рынке, а также другую продукцию, на которую может быть оказано влияние |
3.2.3. Система внесения изменений
Внедрение новых решений, постоянное улучшение, результаты контроля процесса и качества продукции и САРА приводят к изменениям. Для оценки, согласования и реализации изменений надлежащим образом компания должна располагать эффективной системой внесения изменений. Существует различие между внесением изменений до первоначального представления материалов в надзорные органы и после представления, если в соответствии с региональными требованиями необходимо уведомление надзорного органа. Система внесения изменений обеспечивает постоянное улучшение своевременно и эффективно. Должна быть обеспечена высокая степень гарантии того, что изменение не приведет к непредвиденным последствиям. Система внесения изменений должна включать следующие элементы с учетом этапа жизненного цикла:- Анализ риска для качества, который выполняется для оценки предлагаемых изменений. Затраты труда, степень формализации и документального оформления при проведении анализа должна соответствовать степени риска;
- Оценку предлагаемых изменений с учетом требований лицензии, включая параметры для разработки, которые были представлены, и/или текущее понимание продукта и процесса. Следует оценить, необходимо ли уведомление надзорного органа в соответствии с региональными требованиями. Согласно ICH Q8 работа в пределах параметров для разработки (установленных параметров) не является изменением (в отношении уведомления надзорного органа). Однако, с точки зрения фармацевтической системы качества все изменения следует оценивать в соответствии с системой внесения изменений компании;
- Следует выполнить оценку предлагаемых изменений группами экспертов, имеющими опыт и знания в соответствующей области (например, фармацевтической разработки, производства, качества, работы с надзорными органами и медицины) для подтверждения технической обоснованности изменений. Следует установить критерии оценки предлагаемого изменения в перспективе;
- Следует выполнить оценку изменений после их реализации для подтверждения достижения цели, с которой вносилось изменение, и отсутствия отрицательного эффекта на качество продукции.
| Фармацевтическая разработка | Передача технологии | Коммерческое производство | Прекращение производства |
| Изменение является естественной частью процесса развития и должно быть оформлено документально; степень формализации процесса внесения изменений должна соответствовать стадии фармацевтической разработки | Система внесения изменений должна обеспечивать организацию выполнения и документальное оформление наладки процесса при переносе технологии | Коммерческое производство должно иметь формальную систему внесения изменений. Надзор со стороны подразделения качества должен дать уверенность в том, что применены научный подход и методы оценки риска | Любые изменения после прекращения производства продукции должны проводиться через соответствующую систему внесения изменений |
3.2.4. Рассмотрение процесса и качества продукции
Целью рассмотрения процесса и качества продукции является обеспечение их соответствия требованиям в течение жизненного цикла. В зависимости от размеров и сложности компании оно может быть разделено на несколько частей для разных уровней управления и должно предусматривать своевременную и эффективную связь и развитие процесса для доведения необходимых вопросов по качеству до сведения руководства высших уровней.- Рассмотрение должно включать в себя:
- Результаты инспектирования надзорными органами и наблюдения, результаты аудитов и оценки, а также обязательства, направленные в надзорные органы;
- Периодическое рассмотрение качества, которое может включать в себя:
- Реакция потребителей, например, рекламации и отзывы продукции;
- Заключения о работе процесса и контроле качества продукции;
- Эффективность изменений процесса и продукции, включая внесенные в порядке корректирующих и предупреждающих действия;
- По результатам рассмотрения следует определить необходимые действия, например:
- Улучшения технологического процесса и продукции;
- Необходимое обеспечение, обучение или реорганизация;
- Приобретение и распространение знаний.
| Фармацевтическая разработка | Передача технологии | Коммерческое производство | Прекращение производства |
| Результаты рассмотрения могут быть применены к обеспечению выполнения требований к продукции и разработке процесса | Рассмотрение должно способствовать возможности производства продукции и увеличению объема выпуска | Рассмотрение должно представлять собой структурированную систему, как указано выше, и должно способствовать постоянному улучшению | Рассмотрение должно включать в себя такие элементы как стабильность продукции и рекламации на качество продукции |
4. Постоянное улучшение фармацевтической системы качества
В этом разделе приводятся действия, которые должны выполнятся для организации постоянного улучшения фармацевтической системы качества.4.1. Рассмотрение фармацевтической системы качества
Руководство предприятия должно располагать формальным процессом рассмотрения фармацевтической системы качества на периодической основе. Этот процесс должен включать в себя:- Оценку достижения целей фармацевтической системы качества;
- Оценку показателей, которые могут быть использованы для контроля эффективности процесса внутри фармацевтической системы качества, например:
- Рекламации, отклонения, СAPA и внесение изменений;
- Обратная связь от работ, выполняемых по контракту;
- Внутренняя оценка процессов, включая анализ рисков, анализ тенденций и аудиты;
- Внешняя оценка, такая как инспекции надзорными органами, наблюдения и аудиты заказчиками.
4.2. Контроль внутренних и внешних факторов, влияющих на фармацевтическую систему качества
К факторам, подлежащим контролю со стороны руководства, могут относиться:- Проекты нормативных правовых актов, руководств документов, относящихся качеству, которые могут оказать влияние на фармацевтическую систему качества;
- Обратная связь от работ, выполняемых по контракту;
- Новые решения, которые могут улучшить фармацевтическую систему качества;
- Изменения в праве собственности.
4.3. Результаты рассмотрения и контроля
Результаты рассмотрения руководством фармацевтической системы качества контроля внешних и внутренних факторов могут включать в себя:- Улучшение фармацевтической системы качества и связанных с нею процессов;
- Размещение или перемещение ресурсов и/или обучение персонала;
- Пересмотр политики в области качества и целей качества;
- Документальное оформление, своевременное и эффективное оповещение о результатах рассмотрения и действиях, включая доведение необходимой информации до высшего руководства.
5. Словарь
В ICH Q10 используются определения по ICH и ИСО, если они установлены. Для целей ICH Q10 слова «требование», «требования» или «необходимо» даны согласно определениям ИСО и не обязательно отражают требования надзорных органов (требования нормативных правовых документов – прим. переводчика). Источник определения указан в скобках после определения. Если определение в ICH или в ИСО отсутствуют, то дается определение ICH Q10. Прим. переводчика: В данном тексте это условие выполняется не всегда, поскольку переводы определений на русский язык в некоторых стандартах ИСО не отражают смысла термина. Возможности процесса (Capability of a Process): Способность процесса производить продукцию, которая будет соответствовать требованиям к этой продукции. Возможности процесса могут быть также определены в статистических терминах. [ГОСТ ISO 9000–2011] Внесение изменений (Change Management): Системный подход к формированию предложений, оценке, согласованию, внедрению и пересмотру изменений. [ICH Q10] Постоянное улучшение (совершенствование) (Continual Improvement): Повторяющаяся деятельность по увеличению способности выполнять требования. [ГОСТ ISO 9000–2011] Стратегия контроля (Control Strategy): Плановый комплекс мер по контролю, построенный исходя из понимания продукта и процесса в текущее время, обеспечивающий выполнение требований к процессу и качеству продукции. Эти меры могут включать контроль параметров и свойств, относящихся к субстанциям лекарственных средств, материалам и компонентам лекарственных средств, условий работы помещений и оборудования, внутрипроизводственный контроль, спецификации на готовую продукцию и связанные с ними методы и периодичность контроля. [ICH Q10] Корректирующее действие (Corrective Action): Действие, предпринятое для устранения причины обнаруженного несоответствия или другой нежелательной ситуации. П р и м е ч а н и е: Корректирующее действие предпринимают для предотвращения повторного возникновения события, а предупреждающее действие – для предотвращения возникновения события. [ГОСТ ISO 9000–2011] Параметры для разработки (Design Space): Многомерная комбинация и взаимодействие переменных (например, свойств материалов) и параметров процесса, которые обеспечивают выполнение требований к качеству. [ICH Q8] Средство реализации (Enabler): Инструмент или процесс, который позволяет достичь цели. [ICH Q10] Обратная связь/Предпринимаемые действия (Feedback/Feedforward): Обратная связь (Feedback): Изменение или контроль процесса на основе его результата или эффекта. Предпринимаемые действия (Feedforward): Изменение или контроль процесса на основе предполагаемого результата или эффекта. [Oxford Dictionary of English. Oxford Press; 2003] Обратная связь/Предпринимаемые действия могут применяться в техническом плане в стратегии контроля процесса и, в принципе, обеспечении качества. [ICH Q10] Инновация (Innovation): Внедрение новых технологий или методов. [ICH Q10] Обеспечение информацией (Knowledge Management): Систематический подход к получению, анализу, хранению и распространению информации о продукции, технологических процессах и компонентах [ICH Q10] Работы по контракту (Outsources Activities): Работы, выполняемые исполнителем в соответствии с письменным соглашением с заказчиком. [ICH Q10] Показатели работы (Performance Indicators): Количественные величины, используемые для оценки качества и отражающие работу организации, процесса или системы, также именуемые «параметры работы – performance metrics» в некоторых регионах. [ICH Q10] Фармацевтическая система качества (Pharmaceutical Quality System): Система организации, которая направляет и контролирует работу фармацевтической компании в отношении качества. [ICH Q10 на основе ИСО 9000:2005] Предупреждающее действие (Preventive Action): Действие, предпринятое для устранения причины потенциального несоответствия или другой нежелательной ситуации. Примечание: Предупреждающее действие предпринимают для предотвращения воз- никновения события, а корректирующее действие – для предотвращения повторного возникнове- ния события. [ГОСТ ИСО 9000–2011] Выпуск продукта (Product Realisation): Получение продукта с показателями качества, соответствующими нуждам пациентов, специалистов в области здравоохранения и надзорных органов (включая соответствие лицензии) и внутренним требованиям заказчиков. [ICH Q10] Качество (Quality): Степень соответствия комплекса свойств продукции, систем или процесса требованиям. [ICH Q9] Руководство по качеству (Quality Manual): Документ, определяющий систему менеджмента (обеспечения) качества организации. [ГОСТ ИСО 9000–2011] Задачи качества (Quality Objectives): Средства реализации требований политики качества и стратегий в действия, которые могут быть оценены. [ICH Q10] Планирование качества (Quality Planning): Часть системы менеджмента (обеспечения) качества, направленная на установление целей в области качества и определяющая необходимые операционные процессы и соответствующие ресурсы для достижения целей качества. [ГОСТ ИСО 9000–2011] Политика в области качества (Quality Policy): Общие намерения и направления деятельности организации в области качества, официально сформулированные высшим руководством. [ГОСТ ИСО 9000–2011] Анализ рисков (Quality Risk Management): Систематический процесс оценки, контроля и рассмотрения рисков для качества лекарственных средств в течение жизненного цикла. [ICH Q9] Высшее руководство (Senior Management): Лицо (лица), которое управляет и контролирует компанию или производство на высшем уровне, имеет полномочия и не- сет ответственность за использование ресурсов в пределах компании или производства. [ICH Q10 на основе ИСО 9000:2005] Состояние контроля (State of Control): Условия, при которых комплекс мер по контролю последовательно обеспечивает выполнение процесса и требований к качеству. [ICH Q10]Приложение 1
Возможности усиления научного и основанного на анализе рисков подходов в работе надзорного (регулирующего) органа Примечание – Это приложение отражает потенциальные возможности для совершенствования нормативных подходов Конкретный процесс регулирования определяется региональными нормами.| Наименование | Возможности |
| 1. Соответствие GMP | Соответствие – должно быть |
| 2. Демонстрация эффективности фармацевтической системы качества, включая эффективное использование принципов анализа рисков (например, по ICH Q9 и ICH Q10) | Возможности для:
|
| 3. Демонстрация понимания продукта и процесса, включая эффективное использование принципов анализа рисков (например, по ICH Q8 и ICH Q9) | Возможности для:
|
| 4. Демонстрация эффективности фармацевтической системы качества и понимания продукта и процесса, включая использование принципов анализа рисков (например, по ICH Q8, ICH Q9 и ICH Q10) | Возможности для:
|
Приложение 2
Схема модели фармацевтической системы качества по ICH Q10 Схема показывает основные особенности фармацевтической системы качества (PQS), которая охватывает весь жизненный цикл продукта, включая разработку, передачу технологии, коммерческое производство и прекращение выпуска продукции (верхняя часть схемы). PQS дополняет региональные правила GMP, как показано на схеме. Из схемы также видно, что региональные правила GMP применяются к производству препаратов для исследований. Следующая горизонтальная строка показывают важность ответственности руководителей согласно разделу 2 на всех стадиях жизненного цикла продукции. В следующей строке перечисляются элементы PQS, образующие основу модели PQS. Эти элементы применяются соответственно и пропорционально к каждой стадии жизненного цикла с учетом возможностей непрерывного улучшения (совершенствования). Нижние строки показывают средства реализации: обеспечение информацией и анализ рисков, которые применимы ко всем стадиям жизненного цикла. Они способствуют достижению целей PQS в выпуске продукции, контроле и постоянном улучшении.Введение
Принципы анализа рисков успешно используются во многих областях бизнеса и работе правительственных организаций, включая финансы, страхование, профессиональной безопасности, обеспечения здоровья, безопасности в фармации, а также органами надзора в этих областях. Несмотря на то, что есть некоторые примеры использования методов анализа рисков в фармацевтической промышленности, они носят ограниченный характер и охватывают все средства, относящиеся к анализу рисков. Более того, в фармацевтической промышленности большую роль играют системы качества и становится очевидным, что анализ рисков является важным элементом эффективности системы качества. Под риском понимается комбинация вероятности ущерба и тяжести этого ущерба. Однако достичь понимания о применении анализа риска между различны- ми участвующими сторонами трудно, поскольку каждая сторона может иметь свое понимание потенциального ущерба, приписывать различную вероятность и тяжесть различным видам ущерба. Несмотря на то, что к лекарственным средствам имеют отношение различные стороны, включая потребителей, врачей, представителей власти и промышленности, важнейшим фактором является защита потребителя путем снижения риска для ка- чества. В производстве и применении лекарственных средств и их компонентов всегда присутствует некоторая степень риска. Риск для качества продукции является лишь одной из составляющих общего риска. Важно понимать, что качество продукции должно обеспечиваться в течение всего цикла жизни продукции так, чтобы основ- ные свойства лекарственного средства оставались теми же, что были при клиниче- ских испытаниях. Эффективное применение метода анализа рисков может обеспе- чить высокое качество лекарственного средства, потребляемого пациентом, за счет предупредительных мер по обнаружению и устранению потенциальной угрозы для качества при разработке и производстве. Более того, использование методов анализа рисков позволяет принимать лучшие и обоснованные решения, может дать надзор- ным органам более веские основания о способности предприятия справиться с по- тенциальными рисками и может снизить вероятность пропуска упущений надзор- ным органом с учетом продолжительности и серьезности этих упущений. Настоящий документ дает систематизированный подход к анализу риска для качества. Он является отправным документом, независимым от других документов ICH по качеству, но поддерживающих стандарты и правила для фармацевтической промышленности или надзорных органов. Он дает специализированное руководство по принципам и средствам анализа риска для качества, которые могут способство- вать принятию более эффективных и последовательных решений как для надзорных органов, та и для промышленности в отношении субстанций и готовых лекарствен- ных средств в течение их цикла жизни. Оно не вносит нового в существующие требования, устанавливаемые надзорными органами.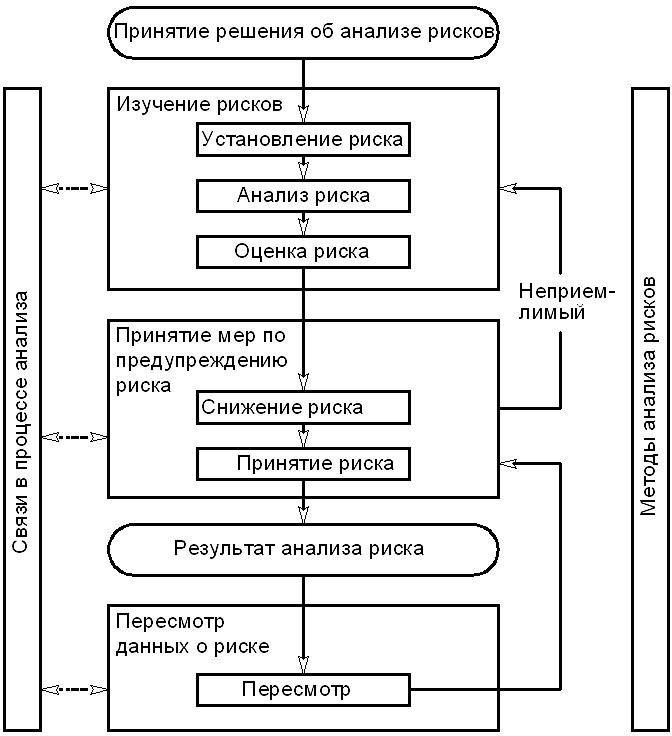 Рис. 1 Типовая схема процесса анализа рисков
Использование формальных методов анализа рисков (общепринятых методов и/или внутренних инструкций) не всегда целесообразно и не всегда необходимо. Допускается использовать неформальные методы на основе эмпирических методов или внутренних инструкций. Применение методов анализа рисков может облегчить выполнение, но не снимает обязанности производителя соответствовать норматив- ным требованиям и не заменяет необходимого взаимодействия между производителем и надзорными органами.
Рис. 1 Типовая схема процесса анализа рисков
Использование формальных методов анализа рисков (общепринятых методов и/или внутренних инструкций) не всегда целесообразно и не всегда необходимо. Допускается использовать неформальные методы на основе эмпирических методов или внутренних инструкций. Применение методов анализа рисков может облегчить выполнение, но не снимает обязанности производителя соответствовать норматив- ным требованиям и не заменяет необходимого взаимодействия между производителем и надзорными органами.
Область применения
Настоящее руководство устанавливает принципы и дает примеры использования методов анализа рисков в применении к различным этапам обеспечения качества лекарственных средств. К этим этапам относятся разработка, производство, распределение (поставка), инспектирование и внесение изменений в процессы на протяжении всего жизненного цикла производства субстанций, готовых лекарственных средств, продуктов биологического и биотехнологического происхождения, включая использование сырья, растворителей, наполнителей, упаковочных материалов и материалов для маркировки.Принципы анализа рисков
Существует два основных принципа анализа рисков:- Оценка риска для качества должна основываться на научном подходе и направлена на защиту потребителя;
- Объем работы и подробность документального оформления при анализе рисков должны определяться степенью риска.
Основное содержание процесса анализа рисков
Анализ рисков является систематизированным процессом оценки, контроля, установления связей и пересмотров, связанных с риском для качества лекарственного средства в течение всего жизненного цикла. Схема анализа рисков показана на рис. 1. Могут использоваться и другие схемы. Значение каждого элемента схемы может отличаться для разных случаев, но каждый из них должен рассматриваться в той степени, которая определяется спецификой конкретного риска. На схеме не показаны точки принятия решений, поскольку решение может быть принято в любой точке процесса. Решениями могут быть: возврат к предыдущему шагу и поиск новой информации, корректировка схемы анализа риска и даже завершение анализа риска, основываясь на полученных данных. Примечание: Слово «неприемлемый» на схеме относится не только к нормативным или правовым требованиям, но также означает повторное проведение анализа рисков.4.1 Ответственность
Анализ рисков, как правило, но не всегда, проводится группой специалистов различного профиля (например, службы качества, развития предприятия, производства, сбыта, маркетинга, инженерных, юридических служб, подразделений статистики и клинических испытаний) в дополнение лицам, имеющим подготовку в области анализа риска. Лица, принимающие решения, должны:- нести ответственность за координацию работы по анализу рисков различными подразделениями предприятия;
- обеспечить правильную постановку задачи анализа рисков, организовать работы по ее решению и привлечению необходимых ресурсов.
4.2 Начало работы по анализу рисков
Анализ риска включает в себя систематизированный процесс координации, облегчение и улучшение принятия решений в отношении рисков. Возможные действия при подготовке работы по анализу рисков включает следующее:- Формирование проблемы и/или вопросов, предположений, связанных с рисками;
- Сбор исходных данных и/или данных о потенциальной опасности, ущербе или воздействии на здоровье человека, связанного с риском;
- Назначение руководителя группы и привлечение необходимых ресурсов;
- Установление сроков выполнения работы, лиц, которым должны быть представлены ее результаты и уровень лиц, принимающих решение в отношении анализа рисков.
4.3 Изучение рисков
Изучение рисков включает в себя установление опасностей, анализ и оценку рисков, связанных с этими опасностями (см. ниже). Изучение риска начинается с ясного описания проблемы или вопросов, связанных с риском. На основе этого определяется метод анализа риска (см. пример в разделе 5) и необходимая информация. Для ясного определения рисков полезно ответить на три основных вопроса:- Что может действовать неправильно (выйти за допустимые пределы)?
- Какова вероятность этого?
- Каковы последствия (тяжесть) этого?
4.4 Меры по предупреждению риска
Меры по предупреждению риска включает в себя принятие решения о снижении и/или принятия данного значения риска. Целью предупреждения риска является его снижение до допустимого уровня. Затраты, связанные с принятием мер, должны быть пропорциональны степени риска. Лица, принимающие решения, могут использовать различные методы, включая анализ «полезный эффект – затраты», для понимания целесообразных мер по предупреждению риска. При принятии мер по предупреждению риска следует ответить на вопросы:- Выходит ли риск за допустимые пределы?
- Что может быть сделано для снижения или устранения риска?
- Каков приемлемый баланс между полезным эффектом, затратами и требуемыми ресурсами?
- Не появляются ли новые риски в результате принятия мер по уже установленным рискам?
4.5 Связи в процессе анализа рисков
Связи в процессе анализа рисков состоит в обмене информацией между лица- ми, принимающими решения и другими участниками работы. Стороны, которые могут участвовать в этом, показаны на рис. 1 стрелками с пунктиром. Связи устанавливаются между причастными сторонами, например, между надзорными органами и производителем, производителем и потребителем, внутри предприятия и пр. Пере- даваемая информация может относиться к существованию, природе, форме, вероятности, тяжести, приемлемости, мерам по предупреждению, устранению, обнаружению рисков или другим факторам. Необязательно устанавливать для каждого случая риска. Связь по принятию решений между промышленностью и надзорными органа- ми может осуществляться в установленном действующими документами порядке.4.6 Рассмотрение данных о риске
Анализ рисков должен стать отправной точкой в процессе обеспечения качества. Следует ввести порядок рассмотрения или контроля событий. Результаты анализа рисков следует рассматривать, чтобы учесть новые данные науки и практики. После начала процесса анализа рисков, его следует применять для случаев, которые могут оказать влияние на решения по анализу рисков для качества, независимо от того, являются ли эти решения плановыми (например, результаты анализа продукции, инспекции, аудитов, контроля измерений) или незапланированными (например, обнаружение причины при исследовании отклонений, отзывы продукции). Периодичность проведения рассмотрения зависит от степени риска. При рассмотрении рисков следует учитывать решения по допустимости риска (п. 4.4).Методы анализа риска
Анализ рисков для качества дает научную и практическую поддержку при принятии решений. Он дает документально оформленные, прозрачные и воспроизводимые методы для выполнения процесса анализа рисков, основываясь имеющихся знаниях об оценке вероятности, тяжести и в отдельных случаях возможности обнаружения риска. Оценка риска для качества выполнялась обычно различными неформальными методами (эмпирическими и/или по внутризаводским инструкциям), основываясь на основе, например, на наблюдениях, анализе тенденций или другой информации. Некоторые подходы по-прежнему позволяют получить полезную информацию на основе анализа рекламаций, дефектов качества, отклонений и наличием средств. В дополнение к этому в фармацевтической промышленности и в надзорных органах могут использоваться известные методы анализа рисков и инструкции предприятий. Ниже приведен неполный список этих методов (см. приложение А и раздел 8):- Основные методы анализа рисков (диаграммы потоков, контрольные листы и пр.);
- Анализ вида и влияния отказов (Failure Mode Effects analysis – FMEA);
- Анализа вида, влияния и критичности отказов (Failure Mode, Effects and Crit- icality Analysis – FMECA);
- Анализ дерева отказов (Fault Tree Analysis - FTA);
- Анализ опасностей в критических контрольных точках (Hazard Analysis and Critical Control Points - HACCP);
- Анализ опасностей для эксплуатации (Hazard Operability Analysis - HAZOP);
- Предварительный анализ опасности (Preliminary Hazard Analysis - PHA);
- Ранжирование и отсеивание рисков (Risk ranking and filtering);
- Вспомогательные статистические методы (Supporting statistical tools).
Интеграция методов анализа риска в промышленность и систему надзора
Анализ риска для качества, будучи введенным в систему обеспечения качества, способствует принятию практичных и обоснованных решений (приложение II). Как было подчеркнуто во введении, применение методов анализа рисков не снижает обязательств предприятий выполнять установленные требования. Однако эффективное применение методов анализа рисков может способствовать принятию более обоснованных решений, обеспечить надзорные органы большей уверенностью в способности предприятия справиться с потенциальными рисками и может влиять на объем и уровень незамеченных надзорным органом упущений. В дополнение к этому анализ рисков позволяет лучше использовать ресурсы всех сторон. Обучение работников предприятий и надзорных органов методам анализа рисков позволяет лучше понять процесс принятия решений и закладывает уверенность в результатах работы по анализу рисков. Методы анализа рисков должны быть включены в существующую практику и документы. В приложении II приведены примеры использования методов анализа рисков в производстве лекарственных средств. Эти примеры даны только для иллюстрации и не являются исчерпывающими или к чему-то обязывающими. Эти приме- ры не вводят новых требований по сравнению с существующими нормативными документами. Примеры для предприятий и надзорных органов (приложение II):- Обеспечение качества.
- Развитие,
- Помещения, оборудование и технологические среды,
- Обращение с материалами,
- Производство,
- Контрольные лаборатории и контроль стабильности,
- Упаковка и маркировка.
- Инспектирование и оценка деятельности.
Определения
Лицо, принимающее решение (Decision maker): Лицо, компетентное и имеющее право принятия соответствующих и своевременных решений в отношении анализа рисков. Выявляемость (Detectability): Способность к обнаружению опасности. Ущерб (Harm): Нанесение вреда здоровью, в том числе из-за утраты лекарственным средством своих свойств или отсутствия лекарственного средства в наличии. Опасность (Hazard): потенциальный источник ущерба (руководство ИСО/МЭК 51). Жизненный цикл продукта (Product Lifecycle): все этапы жизни продукта от начала разработки, включая поставку на рынок, до прекращения существования продукта. Качество (Quality): степень соответствия свойств продукта, системы или процесса заданным требованиям (см. определение качества в ICH Q6a для субстанций и лекарственных средств). См. также определение качества в ГОСТ Р 52537-2006. Анализ риска для качества (Quality risk management): систематизированный процесс оценки, принятия мер, связи и пересмотра в отношении качества лекарственного средства в течение его жизненного цикла. Система обеспечения качества (Quality system): совокупность всех факто- ров, обеспечивающих реализацию политики в области качества и направленных на соответствие требованиям качества. См. также определение системы обеспечения качества в ГОСТ Р 52537-2006. Требования (Requirements): Ясные и полные потребности или ожидания потребителей или лиц, представляющих их интересы (например, медицинских работников, служащих надзорных органов или законодателей). В данном документе тер- мин «требования» относится не только к требованиям закона, правовых или нормативных документов, но и указанные выше потребности или ожидания. Риск (Risk): Комбинация вероятности нанесения вреда и тяжести этого вреда (Руководство ИСО/МЭК 51). Допущение риска (Risk acceptance): решение о допустимости риска (Руководство ИСО/МЭК 73). Анализ риска (Risk analysis): Оценка риска, связанная с данной опасностью. Оценка риска (Risk assessment): Систематизированный процесс сбора информации для принятия решения в ходе анализа риска. Он включает в себя установление опасностей, анализ и оценку рисков, связанных этими опасностями. Связь в процессе анализа рисков (Risk communication): Обмен информацией о риске и анализе риска между лицом, принимающим решение и другими сторонами. Принятие мер по предупреждению риска (Risk control): Действия, направленные на реализацию решений по результатам анализа риска. Оценка риска (Risk evaluation): Сравнение полученных данных о риске с за- данными критериями риска, используя количественные или качественные методы определения значимости риска. Установление риска (Risk identification): Систематическое проведение анализа информации с целью обнаружения потенциальных источников ущерба (опасности) для данного вида риска или проблемы. Работа в области рисков (Risk management): систематическое применение принципов обеспечения качества, инструкций и руководств для оценки, принятия мер, связи и пересмотра данных о риске. Снижение риска (Risk reduction): Действия, направленные на снижение вероятности наступления ущерба и тяжести этого ущерба. Пересмотр данных о риске (Risk review): пересмотр или контроль результатов работы в области рисков с учетом новых разработок (если они есть) и опыта в области анализа риска. Тяжесть (Severity): Мера возможных последствий опасности. Сторона (Stakeholder): лицо, группа лиц или организация, которая может влиять на риск, или быть подверженной влиянию его последствий. Лица, принимающие решения также могут являться сторонами. Для целей настоящего руководства первичными сторонами являются потребители, медицинские работники, над- зорные органы и предприятия. Тенденция (Trend): статистический термин, относящийся к направлению и интенсивности изменений переменных величин.Библиография
- ICH Q8 Pharmaceutical development;
- ISO/IEC Guide 73:2002 – Risk Management – Vocabulary – Guidelines for use in Standards;
- ISO/IEC Guide 51:1999 – Safety Aspects – Guideline for the inclusions in standards;
- Process Mapping by the American Productivity & Quality Center 2002, ISBN1928593739;
- IEC 61025 – Fault Tree Analysis (FTA);
- IEC 60812 Analysis Techniques for system reliability – Procedures for failure mode and effects analysis (FMEA);
- Failure Mode and Effect Analysis, FMEA from Theory to Execution, 2nd Edition 2003, D. H. Stamatis, ISBN 0873895983;
- Guidelines for Failure Modes and Effects Analysis (FMEA) for Medical Devices, 2003 Dyadem Press ISBN 0849319102;
- The Basis of FMEA, Robin McDermott, Raymond J. Mikulak, Michel R. Beaure- gard 1996 ISBN 0527763209;
- WHO Technical Report Series No 908, 2003 Annex 7 Application of Hazard Analysis and Critical Control Points (HACCP) methodology to pharmaceuticals;
- IEC 61882 – Hazard Operability Analysis (HAZOP);
- ISO 14971:2000 – Application of Risk Management to Medical Devices;
- ISO 7870:1993 – Control Charts;
- ISO 7871:1997 – Cumulative Sum Charts;
- ISO 7966:1993 – Acceptance Control Charts;
- ISO 8258:1991 – Shewhart Control Charts;
- What is Total Quality Control?; The Japanese Way, Kaoru Ishikawa (Translated by David J. Liu, 1985, ISBN 0139524339.
Приложение I. Методы анализа рисков
Данное приложение дает общий обзор и ссылки на некоторые методы, которые могут использоваться при анализе рисков предприятиями и надзорными органами. Использование ссылок позволяет получить более подробную информацию о рассматриваемых методах. Приводимые методы не являются исчерпывающими. Важно иметь в виду, что ни один метод или набор методов не является универсальным для анализа рисков.I.1 Основные методы анализа рисков
К простым методам, обычно используемым систематизации данных, структурирования работы и облегчения принятия решений являются:- диаграммы потоков,
- контрольные листы,
- карты процесса,
- диаграммы причин следствий, называемых также диаграммами Исикава или «рыбья кость».
I Анализ вида и влияния отказов (FMEA)
Метод анализа вида и влияния отказов (FMEA, IEC 60812) служит для оценки возможных причин отказов в процессах и их вероятного влияния на результат работы и/или продукт. После того, как установлен вид отказа, снижение риска может использоваться для устранения, ограничения, уменьшения или другого принятия мер в отношении отказов. FMEA основывается на знании продукта и процесса. FMEA дает методику разделения сложного процесса на приемлемые для анализа части. Он является эффективным средством для получения итоговой информации о существенных характеристиках отказов, причин этих отказов и возможных последствиях этих отказов.Возможные области применения
FMEA может использоваться для установления градации рисков и контроля эффективности действий по их снижению. FMEA может применяться к оборудованию и помещениям, анализу производственных операций и их влиянию на продукт или процесс. Он устанавливает эле- менты и операции внутри системы, которые делают ее уязвимой. Результаты FMEA могут использоваться в качестве основы для проектирования (конструирования), дальнейшего анализа или использоваться при дальнейшем развитии.I.3 Анализа вида, влияния и критичности отказов
Область применения FMEA может расширена и включать изучение степени тяжести последствий отказов, вероятности их появления и возможности обнаружения. Метод, сочетающий эти особенности, называется методом анализа характера, влияния и критичности отказов (FMECA, IEC 60812). Для выполнения такого анализа необходимы спецификации (технические условия, технологический регламент) на продукт или процесс. FMECA позволяет определить точки, в которых требуется проведение предупредительных действий с целью сведения рисков к минимуму.Возможные области применения
Применение FMECA в производстве лекарственных средств должно, главным образом, распространяться на отказы и риски, связанные с технологическими процессами, но это не ограничивает область применения этого метода. Результатом использования FMECA является присвоение количественного показателя (весового коэффициента) для каждого вида отказов, что позволяет ранжировать виды отказы с учетом их относительного риска.I.4 Анализ дерева отказов
Метод анализа дерева отказов (FTA, IEC 61025) рассматривает отказы в работе продукта или процесса. Этот метод рассматривает отдельные отказы, но может использоваться и при сочетании многих причин отказов путем построения причинных цепей. Результаты анализа представляются наглядно для форме дерева видов отказов. На каждом уровне дерева дается описание видов отказов с использованием логических символов (и, или и пр.). FTA основывается на понимании процесса экспертами для установления причин отказов.Возможные области применения
Метод FTA может использоваться для построения пути, ведущего к изначальной причине отказа. Он может использоваться для расследования рекламаций и отклонений для полного понимания их изначальной причины и обеспечения уверенности в том, что принимаемые меры полностью устранят эту причину и не приведут к другим последствиям (т. е решение одной проблемы не приведет к появлению новой). FTA является эффективным методом оценки влияния множественных факторов. К результатам работы по FTA относится и наглядное представление причин отказов. Оно полезно как для оценки рисков, так и для разработки программы контроля.I.5 Анализ опасностей в критических контрольных точках
Метод анализа опасностей в критических контрольных точках (Hazard Analysis and Critical Control Points - HACCP) является систематизированным, направленным на предупреждение рисков методом для обеспечения качества, надежности и безопасности продукта (WHO Technical Report series No 908, 2003, Annex 7). Он является структурированным подходом, который использует принципы науки и техники для анализа, оценки, предупреждения и принятия мер в отношении риска или отрицательных последствий опасностей, вызванных проектом (конструкцией), разработкой, производством и использованием продукта. Применение HACCP включает в себя следующие семь этапов:- выполнение анализа опасностей и определение предупредительных мер для каждой операции процесса;
- определение критических контрольных точек;
- установление критических пределов;
- установление системы контроля в критических контрольных точках;
- определение корректирующих действий, которые следует предпринять, когда система контроля показывает, что параметры в критической точке вышли за установленные пределы;
- введение системы, которая подтверждает эффективность работы по НАССР;
- создание системы ведения документации.
Возможные области применения
Метод НАССР следует использовать анализа и принятия мер в отношении рисков, связанных с физической, химической или биологической опасностью включая микробное загрязнение. Этот метод наиболее эффективен, когда есть достаточно подробные данные о продукте или процессе, чтобы определить критические контрольные точки. Результатом анализа является информация о рисках, которая не только позволяет контролировать критические точки в процессе производства, но и на других этапах жизненного цикла.I.6 Анализ опасностей для эксплуатации
Метод анализ опасностей для эксплуатации (Hazard Operability Analysis – HAZOP, IEC 61882) основан на предположении, что риском вызывается отклонениями от проекта (конструкции) или правил эксплуатации. Он является систематизированным методом мозгового штурма для установления опасностей с использованием ключевых слов (например: нет, более, иные чем, часть чего-то и пр.) в применении к определенным параметрам (например, загрязнение, температура) для облегчения установления потенциальных отклонений от нормальной эксплуатации или проекта (конструкции). Для работы по этому методу во многих случаях привлекаются группы специалистов, имеющие опыт в разработке процессов или продукции и их применении.Возможные области применения
Метод HAZOP может применяться для производственных процессов, включая использование продукции и материалов, получаемых из внешних источников, а также поставщиков, оборудования и помещений, используемых для производства субстанций и готовых лекарственных средств. Он также использовался, в основном, в фармацевтической промышленности для оценки безопасности процессов. Так же и для НАССР, результатом работы по HAZOP является перечень критических операций для анализа рисков. Это позволяет организовать регулярный контроль в критических контрольных точках производственного процесса.I.7 Предварительный анализ опасности
Метод предварительного анализа опасности (Preliminary Hazard Analysis - PHA) основан на использовании предшествующего опыта или информации об опасности или при невозможности установить будущие опасность, опасные ситуации и события, которые могут привести к ущербу или невозможности дать оценку их вероятности для данного вида деятельности, помещений, оборудования, продукта или системы. Метод состоит в:- установлении существования риска,
- количественной оценке возможного ущерба здоровью,
- относительного ранжирования опасности, используя комбинацию тяжести и вероятности нанесения ущерба,
- определение возможных способов устранения риска.
Возможные области применения
Метод PHA может применяться для систем и выделения приоритетов по опасностям, когда отсутствует возможность использования более информативных методов. Он может использоваться для оценки проекта помещений, конструкции оборудования и построения процессов, а так же для оценки видов опасностей исходя из характера производства, вида продукции и конкретных данных о продукте. Как правило, метод PHA применяется на ранних стадиях разработки проекта, когда подробная информация о проекте или технологических операциях отсутствует, т. е. метод дает предварительную проработку перед проведением дальнейших исследований. Как правило, опасности, обнаруженные с помощью метода PHA анализируются в дальнейшем другими методами, том числе рассмотренными ранее в настоящем приложении.I.8 Ранжирование и отсеивание рисков
Метод ранжирования и отсеивания рисков (Risk ranking and filtering) используется для сравнения группирования рисков по их значимости (ранжирования). Ран- жирование рисков для сложной системы требует, как правило, анализа многих количественных и качественных факторов для каждого вида риска. Метод разделяет основной вопрос анализа рисков на столько элементов, сколько требуется для учета всех факторов, влияющих на риск. Эти факторы группируются в единую относи- тельную последовательность рисков, которая может использоваться для ранжирования. «Фильтры» в виде весовых коэффициентов или усеченных последовательностей, которые могут использоваться группирования или ранжирования рисков с целью проведения анализа или принятия решений.Возможные области применения
Ранжирование рисков и их отсеивание при планировании проведения инспекций (аудита) производств надзорными органами или предприятиями. Методы ранжирования рисков дают наибольший эффект, когда полученная совокупность рисков и требующих учета факторов характеризуется разнообразием и трудностью анализа с использованием одного метода. Ранжирование рисков целесообразно, когда требуется как количественную, так и качественную характеристику рисков в рамках одной работы.I.9 Вспомогательные статистические методы
Вспомогательные статистические методы (Supporting statistical tools) способствуют принятию решений в отношении рисков для качества. Они позволяют более эффективно выполнять оценку данных, значимости данных и более надежно принимать решения. Перечень наиболее часто применяемых статистических методов в фармацевтической промышленности включает в себя: I Контрольные карты, например:- Карты контроля приемлемости (ИСО 7966),
- Контрольные карты с средними арифметическими значениями и пределами предупреждения (ИСО 7873),
- Кумулятивные карты (ИСО 7871),
- Контрольные карты Шухарта (ИСО 8258),
- Скользящие средние, которым придается определенный вес; II Планирование эксперимента;
- Гистограммы;
- Диаграммы Парето;
- Анализ характеристик процесса.
Приложение II. Области применения методов анализа рисков
Данное приложение предназначено для ориентации работников промышленности и надзорных органов в выборе методов анализа рисков. Следует иметь в виду, что выбор метода полностью определяется конкретной задачей. Приведенные примеры служат только для целей иллюстрации и служат только для информирования потенциальных пользователей.II.1 Анализ рисков как часть системы обеспечения качества
Анализ рисков может использоваться в следующих случаях:Документация:
- при пересмотре текущей документации и работе с нормативными документами;
- при определении предмета и содержания инструкций, руководств и пр.
Обучение и повышение квалификации:
- для оценки соответствия первоначального и повторного обучения образованию, опыту и практических навыков персонала, а также для периодической оценки обучения, в том числе его эффективности;
- для оценки способности персонала (его подготовки, опыта, квалификации и физических качеств) надежно выполнять порученную работу и не допускать отрицательного влияния на качество продукта.
Дефекты качества:
- для создания основы обнаружения и оценки влияния на качество различных дефектов, тенденций, отклонений, исследований, выхода параметров за пределы спецификаций, анализа рекламаций;
- для формирования связей по анализу рисков и разработки мер в отношении существенных дефектов продукции, контактов с надзорными органами и пр.
Проведение инспекций и аудитов
Для определения периодичности и объема аудитов (внешних и внутренних) следует учесть:- Действующие нормативные и правовые документы,
- Общие данные о соответствии предприятия или производства установленным требованиям и историю предприятия;
- Эффективность работы по анализу рисков;
- Сложность производства;
- Сложность технологических процессов;
- Сложность продукции и ее терапевтической эффективности;
- Число и тяжесть дефектов качества;
- Результаты предыдущих инспекций/аудитов;
- Основные изменения в здании, оборудовании, процессах, ключевом персонале;
- Опыт производства продукта (например, частота выпуска серий продукта,их размер, число серий);
- Результаты испытаний, проводимых аккредитованными контрольными лабораториями.
Периодический анализ:
- для выбора, оценки и интерпретации результатов изучения тенденций при анализе качества продукта;
- для интерпретации данных контроля, например, для определения необходимости проведения повторной аттестации (валидации) или внесения изменений в методы отбора проб.
Планирование изменений и контроль изменений:
- для планирования изменений на основе информации, полученной в ходе производства и развития фармации;
- для оценки влияния изменений на готовый продукт;
- для оценки влияния изменений в помещениях, оборудовании, материалах, производственном процессе и внедрении новой техники;
- для определения действий, предшествующих внесению изменений, например, дополнительные испытания, повторная аттестация или взаимодействие с над- зорными органами.
Непрерывное совершенствование
Заключается в непрерывном совершенствовании процессов в течение жизненного цикла продукта.II.2 Анализ рисков как часть работы надзорных органов
Анализ рисков может использоваться в следующих случаях:Инспектирование и оценка соответствия:
- для определения необходимых ресурсов, например, при планировании инспекций, их периодичности и объема (см. раздел «Аудит» в приложении II.1;
- для оценки значимости, например, дефектов качества, возможных отзывов продукции и обнаруженных при проведении инспекции недостатков;
- для принятия решений по результатам проведения инспекции;
- для оценки данных, представляемых предприятиями, включая данные о развитии;
- для оценки влияния предложенных изменений;
- при определении факторов риска, в отношении которых должен быть обмен информацией между инспекцией и экспертами для лучшего понимания того, как риск может быть предупрежден (например, для выпуска продукции по параметрам, применении методов анализа процессов – Process Analytical Technology, PAT).
II.3 Анализ рисков как часть процесса развития
Анализ рисков может использоваться с целью:- разработки продукта высокого качества и производственного процесса для обеспечения непрерывного соответствия продукта своему назначению;
- расширение знаний о характеристиках продукции с помощью различных показателей (например, распределением частиц по размерам, содержанием влаги, текучести), параметров технологического процесса и вариантах его реализации;
- оценки критических свойств исходных материалов, растворителей, активных фармацевтических субстанций, наполнителей и упаковочных материалов;
- разработки спецификаций, в которых указываются критические параметры процесса и устанавливается порядок контроля при производстве (например, с использованием данных о разработке продукта в плане значимости показателей качества для клинической эффективности продукта и возможности их контроля при производстве);
- уменьшения разброса свойств качества:
- сокращения дефектов продукта и материалов;
- сокращения дефектов в производстве;
- оценки необходимости дальнейшего анализа (например, биоэквивалентности, стабильности) с учетом расширения производства и внедрения новых технологий;
- использования принципа «проектируемого пространства» - “design space” (ICH Q8).
II.4 Анализ рисков помещений, оборудования, технологических сред и инженерных сетей
Анализ рисков может использоваться в следующих случаях:Проект (конструкция) помещений и оборудования:
- для выделения необходимых при проектировании зданий и помещений с учетом:
- потоков материалов и персонала,
- минимизации загрязнений,
- борьбы с насекомыми,
- предотвращения перепутывания,
- использования открытых и закрытых систем,
- чистых помещений и изоляторов,
- специализированных или изолированных (отделенных) помещений и оборудования;
- для определения вида материалов оборудования, вступающих в контакт с продуктом и первичной упаковкой,
- для определения необходимых технологических сред и инженерных сетей (например, пар, газы, вода, сжатый воздух, источники энергии, системы отопления, вентиляции и кондиционирования);
- планирование предупредительного технического обслуживания оборудования, например, инвентаризации запасных частей.
Чистота оборудования:
- для защиты продукта от влияния внешних факторов, включая химические, микробиологические и физические загрязнения (опасности), например, разработка требований к одежде, порядку переодевания, гигиене);
- для защиты окружающей среды (например, персонала, объектов, чувствительных к перекрестным загрязнениям) от опасностей, связанных с производством данного продукта.
Аттестация помещений, оборудования, технологических сред и инженерных сетей:
Выполняется для определения рамок и объема работ по аттестации помещений, зданий, производственного оборудования и/или лабораторных приборов, включая поверку (калибровку).Очистка оборудования и контроль окружающей среды:
- для разделения действий и решений, основываясь на показателях назначения оборудования (например, универсальное и специализированное, производство сериями и непрерывное производство);
- для определения пределов приемлемости (заданных пределов) при аттестации процессов очистки.
Поверка (калибровка) и техническое обслуживание
Используется при разработке графиков поверки (калибровки) и технического обслуживания.Компьютерные системы и компьютерное контрольное оборудование:
- для выбора компьютера и программного обеспечения (модульное, структурированное, устойчивое к отказам);
- для определения продолжительности и содержания аттестации (валидации), например:
- установление критических параметров,
- задания требований и выбора конструкции,
- пересмотр системы кодирования;
- длительность испытаний и выбор методов испытаний;
- надежностей электронных записей и электронных кодов.
II.5 Анализ рисков как часть работы с материалами
Анализ рисков может использоваться в следующих случаях:Анализ и оценка поставщиков и исполнителей, работающих по контракту:
Выполняется для обеспечения полноты информации и обоснованности оценки поставщиков и исполнителей, работающих по контракту (например, проведение аудитов, заключение соглашений с поставщиками об обеспечении качества).Исходные материалы:
Выполняется для оценки различий и возможных рисков для качества, связанных с разбросом характеристик исходных материалов (например, за счет старения, методов синтеза).Использование материалов:
Выполняется для:- определения возможности использования материала, находящегося в карантинном хранении (например, в данном производстве);
- определения пригодности к переработке и использования возвращенных материалов.
Хранение, работа складов и условия распределения:
Выполняется для:- оценки соответствия условий хранения и транспортирования (например, температуры, влажности, конструкции упаковки) заданным требованиям;
- определения влияния нарушений в хранении или транспортировании (например, при организации холодовой цепи) с учетом руководств ICH;
- поддержания инфраструктуры (например, способности обеспечивать требования к отгрузке, промежуточного хранения, обращения с опасными материалами и требующими контроля субстанциями, таможенной очистке);
- обеспечения информацией в отношении доступности лекарственных средств, например, с использованием ранжирования рисков в цепи поставки).
II.6 Анализ рисков как часть производства
Анализ рисков может использоваться в следующих случаях:Аттестация (испытания):
Выполняется для:- установления пределов (рамок) и объема аттестации (например, аналитических методов, процессов, оборудования, методов очистки);
- определения объема последующих действий (например, отбора проб, контроля, повторной аттестации);
- установления различий между критическими и некритическими этапами процесса для проведения исследований при аттестации (валидации).
Внутрипроизводственный отбор проб и контроль
Выполняется для:- определения периодичности и объема выполнения внутрипроизводственного контроля (например, обоснования сниженного объема контроля определенных условиях);
- оценки и обоснования использования методов анализа процессов в сочетании с выпуском продукции по параметрам и в реальном времени.
Планирование производства
Предусматривает соответствующие методы планирования (например, выделение производства, производство кампаниями или совпадающими последовательностями производства).II.7 Анализ рисков как часть лабораторного контроля и контроля ста- бильности
Анализ рисков может использоваться в следующих случаях:Выхода за пределы спецификаций
Выполняется для установления возможных исходных причин и разработки корректирующих действий при анализе случаев выхода за пределы спецификаций.Контроля периодичности повторных испытаний и срока годности
Выполняется для оценки правильности хранения и испытаний исходных мате- риалов, наполнителей и промежуточной продукции.II.8 Анализ рисков как часть процессов упаковки и маркировки
Анализ рисков может использоваться для:Конструкции упаковки
Выполняется для конструирования вторичной упаковки с целью защиты продукта в первичной упаковке (например, для того, чтобы удостовериться в подлинности продукта, читаемости маркировки).Выбор системы укупоривания
Выполняется для определения критических параметров системы укупоривания контейнеров (первичной упаковки).Контроль маркировки
Выполняется при разработке инструкций по контролю, основываясь на возможности перепутывания из-за неправильного нанесения маркировки или из-за не- правильных вариантов одной и той же маркировки.1. Введение
1.1 Сайт мастер файл разрабатывается производителем лекарственных средств и должен включать следующую специальную информацию о политике в области обеспечения качества и деятельности предприятия (производственной площадки), производстве и/или контроле качества процесса по выпуску лекарственных средств на данной площадке и любых работах в соседних и близлежащих зданиях, которые следует учесть. Если на данной площадке выполняется только часть операций по производству лекарственных средств, то в сайт мастер файл следует включать только эти операции, например, проведение анализа, упаковку и т. д. 1.2 Для представления в надзорные органы сайт мастер файл должен содержать четкую информацию о деятельности производителя, относящейся к выполнению правил GMP, которые могут быть полезны при общем контроле и эффективном планировании и проведении инспекций на соответствие требованиям GMP. 1.3 Сайт мастер файл должен содержать достаточную информацию, но объем его не должен превышать по возможности 25-30 стр., плюс приложения. Рекомендуется приводить планировочные решения или схемы в простом виде вместо текстового описания. Сайт мастер файл, включая приложения, должен быть читаемым при представлении на листах бумаги формата А4. 1.4 Сайт мастер файл входит в состав документации системы качества производителя и должен содержаться в соответствующем порядке. Сайт мастер файл должен иметь номер издания, срок ввода в действие и дату, до которой он должен быть пересмотрен. Он подлежит регулярному рассмотрению, чтобы убедиться в его соответствии действительности. Каждое приложение может иметь свою дату ввода в действие, допускающее независимый пересмотр.2. Цель
Данные пояснения служат руководством производителю лекарственных средств по подготовке сайт мастер файла, который может быть полезен надзорному органу в планировании и проведении инспекции на соответствие GMP.3. Область применения
Данные пояснения предназначены для применения при подготовке сайт мастер файла и определении его содержания. Необходимость в подготовке сайт мастер файла может определяться другими нормативными правовыми документами. Данные пояснения применимы ко всем видам операций по производству лекарственных средств, таким как производственные операции, упаковка и маркировка, испытания, перемаркировка и переупаковка, всех видов лекарственных средств. Настоящее руководство также может использоваться при подготовке сайт мастер файла или соответствующего документа банками крови и тканей и производителями активных фармацевтических субстанций.4. Содержание сайт мастер файла
Содержание сайт мастер файла приведено в приложении.1. Приложение: Содержание сайт мастер файла
1 Общие данные о производстве1.1 Контактная информация
- Наименование и юридический адрес;
- Наименование и фактический адрес площадки, здания и производств, находящихся на площадке;
- Контактная информация производителя, в т. ч. доступные в течение 24 ч телефонный номер лица для контактов в случае обнаружения дефектов продукции или отзыва;
- Идентификационный номер площадки, например, в системе GPS или другой системе геолокации, номер D-U-N-S (Data Universal Numbering System – Всемирная система нумерования данных) – уникальный идентификационный номер, присваиваемый американской компанией Dun & Bradstreet1
1.2 Разрешенные виды деятельности по производству лекарственных средств наплощадке
- Копия действующей лицензии на производство, выданного соответствующим компетентным органом (Приложение 1 к сайт мастер файлу); или ссылка на базу данных EudraGMP, если это применимо. Если компетентный орган не выдает лицензию на производство, то это должно быть указано.
- Краткие данные о производстве, импорте, экспорте, реализации и других видах деятельности в соответствии с лицензией, выданной компетентным органом, включая зарубежные организации с разрешенными видами деятельности, относящимися к дозированным формам, соответственно не указанные в лицензии на реализацию;
- Виды продукции, выпускаемые в настоящее время на площадке (список с Приложении 2 к сайт мастер файлу), не указанные в Приложении 1 или базе данных EudraGMP;
- Перечень инспекций по GMP, проведенных на площадке в течение последних пяти лет, включая даты, страну и наименование компетентного органа, который проведет инспектирование. Следует включить также копию действующего сертификата GMP (Приложение 3) или ссылку на базу данных EudraGMP, при наличии.
1.3 Любые другие виды деятельности, производимые на площадке
- Описание нефармацевтических видов деятельности, если они есть.
2. Система обеспечения качества производителя
2.1 Данные о системе обеспечения качества
- Краткое описание и ссылки на использованные стандарты;
- Ответственность по поддержанию системы обеспечения качества, включая высшее руководство;
- Информация о разрешенных видах деятельности, включая даты и содержание разрешительных документов и наименования выдавших их органов.
2.2 Порядок выпуска готовой продукции
- Подробное описание требований к квалификации (образованию и опыту работы) уполномоченного лица, отвечающего за выдачу разрешения на вы- пуск серии, и документы по выпуску;
- Общее описание порядка выдачи разрешения на выпуск и процедуры выпуска;
- Функции уполномоченного лица в карантинном хранении и выпуске готовой продукции и оценке соответствия регистрационным документам;
- Взаимоотношения между уполномоченными лицами в случаях, когда действуют несколько уполномоченных лиц;
- Указание на то, применять ли технологию анализа процесса (РАТ) и/или выпуск в реальном времени или выпуск по параметрам.
2.3 Работа с поставщиками и подрядчиками
- Краткое описание о цепи поставки и программе проведения внешних аудитов;
- Краткое описание системы аттестации подрядчиков, производителей активных фармацевтических субстанций (АФС) и поставщиков других критических материалов;
- Меры, принимаемые по обеспечению соответствия выпускаемой продукции руководствам TSE (Transmitting animal spongiform encephalopathy – Передача губчатой энцефалопатии животных);
- Меры, принимаемые в случае обнаружения или подозрения на фальсификацию продукции (например, неупакованные таблетки), активных фармацевтических субстанций или вспомогательных материалов;
- Привлечение организации (лиц) для проведения научных, аналитических или других работ, относящихся к производству или проведению анализов;
- Перечень производств или лабораторий, работающих по контракту с адресами и контактными данными и схемами снабжения для производства и проведения анализов по контракту, например, стерилизации первичных упаковочных материалов для асептических процессов, контроля исходных материалов и т. д. (должны быть представлены в Приложении 4);
- Краткое описание ответственности заказчика и исполнителя работ по контракту в плане соответствия разрешительным документам (если это не сделано во исполнение п. 2.2.).
2.4 Анализ риска
- Краткое описание методов анализа рисков, используемых производителем;
- область применения и основной предмет анализа рисков, включая краткое описание любых действий, выполняемых на уровнях предприятия и подразделений. Следует упомянуть применение анализа рисков для оценки непрерывности поставок.
2.5 Анализ качества продукции
- Краткое описание порядка проведения анализа.
3. Персонал
- Блок-схема организационной структуры с указанием подразделений обеспечения и контроля качества, производства и должностей руководителей, включая руководство предприятия и уполномоченных лиц (Приложение 5);
- Численность работников подразделений обеспечения и контроля качества, производства, складов и сбыта.
4. Помещения и оборудование
4.1 Помещения
- Краткое описание предприятия, размеры площадки и перечень зданий. Если производства продукции для различных рынков (местного, Евросоюза, США и др.) размещены в разных зданиях на площадке, то в перечне должны быть указаны соответствующие рынки (если это не приведено в п. 1.1);
- Упрощенный план или описание производственных зон с указанием масштаба (не требуется приводить архитектурные или инженерные чертежи);
- Планы помещений производства и схемы потоков (Приложение 6) с указанием классов чистых помещений и перепадов давления между соседними зонами и видов операций (например, смешивание, наполнение, хранение, упаковка и т. д.);
- Планы складов и зон хранения с выделенными зонами для хранения и обращения с высокотоксичными, опасными и сенсибилизирующими материалами и их обозначением, если требуется;
- Указание расходов приточного воздуха, температуры, влажности, перепадов давления, кратностей воздухообмена и наличия (отсутствия) рециркуляции с указанием процентного соотношения рециркуляционного воздуха.
- Наименование видов воды
- Схемы подготовки воды (Приложение 7).
4.2 Оборудование
4.2.1 Перечень основного производственного и контрольного (в лаборатории) оборудования с критическими элементами (Приложение 8).Очистка и дезинфекция
- Краткое описание методов очистки и дезинфекции поверхностей, вступающих в контакт с продукцией (ручная очистка, автоматическая система очистки на месте и т. д.).
Критические в плане требований GMP компьютерные системы
- Описание критических компьютерных систем в плане выполнения требований GMP (исключая оборудование со специальными программируемыми логическими контроллерами – Programmable Logic Controllers, PLCs).
5 Документация
- Описание системы документации (например, электронная, ручная);
- Когда хранение (архив) документов и протоколов находится за пределами площадки (включая данные фармаконадзора, когда применимо): перечень видов документов/протоколов; имя и адрес места хранения и оценка времени, требуемого для получения документов из архива за пределами площадки.
6. Продукция
6.1 Наименования продукции
(могут быть сделаны ссылки на Приложения 1 и 2)- Перечень наименований выпускаемой продукции, включая:
- перечень дозированных форм продукции для человека и применения в ветеринарии, выпускаемых на площадке
- перечень дозированных форм лекарственных средств для исследований, выпускаемых для клинических исследований на площадке, и, если их производство отделено от коммерческого производства, – данные о технологических зонах и персонале
- Применяемые токсичные и опасные вещества (например, с высокой фармакологической активностью и/или с сенсибилизирующими свойствами);
- Наименование продукции, производимой в выделенных зонах (на специализированном оборудовании) или циклами, если она есть;
- Данные о технологии анализа процесса (РАТ), если она применяется: общая информация и данные о компьютерных системах.
6.2 Аттестация процесса
- Краткое описание общего подхода;
- Данные, относящиеся к повторной обработке и переработке.
6.3 Материалы и склады
- Порядок обращения с исходными и упаковочными материалами, нерасфасованной и готовой продукцией, включая отбор проб, карантинное хранение, выпуск и хранение на складах;
- Обращение с отклоненными материалами и продукцией.
7. Контроль качества
- Описание системы контроля качества на площадке, включая контроль физических, химических, микробиологических и биологический показателей.8. Реализация, рекламации, дефекты и отзывы продукции
8.1 Реализация (в части, относящейся к производству)
- Виды компаний (лицензированные оптовые поставщики, лицензированные производители и т. д.) и места их нахождения (в Евросоюзе, США и т. д.), которым отгружается продукция с площадки;
- Описание порядка проверки того, что каждый заказчик (получатель) имеет законное право получать лекарственные средства от производителя;
- Краткое описание порядка проверки выполнения требований к окружающей среде при транспортировке, например, поддержания и контроля температуры;
- Порядок реализации и прослеживания продукции;
- Меры по предупреждению попадания продукции от производителя в нелегальную цепь поставок.
8.2 Рекламации, дефекты и отзывы продукции
- Краткое описание порядка работы с рекламациями, дефектами и отзывами продукции.
9. Самоинспекции
- Краткое описание системы проведения самоинспекций с акцентом на критерии выбора зон для плановых инспекций, выполняемых проверок и по- следующих действий.| Приложение 1 | Копия действующего свидетельства о регистрации (лицензии) |
| Приложение 2 | Перечень производимых дозированных форм, включая международные непатентованные наименования (МНН, INN) или общепринятые наименования (при наличии) используемых активных фармацевтических субстанций (АФС) |
| Приложение 3 | Копия действующего сертификата GMP |
| Приложение 4 | Перечень производителей и лабораторий, работающих по контракту, с адресами и данными для контактов и схемами цепей поставок по этим работам |
| Приложение 5 | Организационная структура |
| Приложение 6 | Планы производственных зон, включая пути движения мате- риалов и персонала, и блок-схемы процессов производства каждого вида продукции (дозированной формы) |
| Приложение 7 | Схемы подготовки воды |
| Приложение 8 | Перечень основного технологического и лабораторного оборудования |
Обучение стандартам GMP
Обучение проводится совместно с Ассоциацией инженеров по контролю микрозагрязнений.
Мы проводим семинары в Москве, в том числе с выездом на предприятия по следующим темам:
- Правила GMP (общий курс);
- Специальные разделы правил GMP
- Чистые помещения, с выдачей международного сертификата ICCCS.
- Занятия проводят специалисты с многолетним практическим опытом работы в стране и за рубежом.